

Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling
- PMID: 19726669
- PMCID: PMC2785556
- DOI: 10.1074/jbc.M109.046722
Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling
Abstract
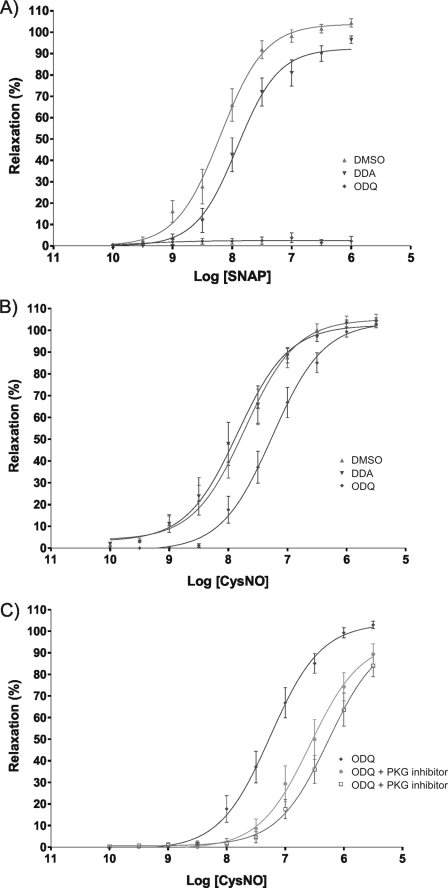
The transnitrosylating nitric oxide (NO) donor nitrocysteine (CysNO) induced a disulfide bond between the two regulatory RI subunits of protein kinase A (PKA). The conventional NO donor S-nitroso-N-acetylpenicillamine failed to do this, consistent with our observation that it also did not promote protein S-nitrosylation. This disulfide oxidation event activated PKA and induced vasorelaxation independently of the classical beta-adrenergic or NO signaling pathway. Activation of PKA had also been anticipated to exert a positive inotropic effect on the myocardium but did not. The lack of positive inotropy was explained by CysNO concomitantly activating protein kinase G (PKG) Ialpha. PKG was found to exert a partial negative inotropic influence regardless of whether PKA was activated by classical beta-receptor stimulation or by disulfide bond formation. This work demonstrates that NO molecules that can induce S-nitrosylation directly activate type I PKA, providing a novel cross-talk to beta-adrenergic-like signaling without receptor or adenylate cyclase stimulation. However, the expected positive inotropic consequences of PKA activation by this novel mechanism are countermanded by the simultaneous dual activation of PKGIalpha, which is also activated by CysNO.
Figures
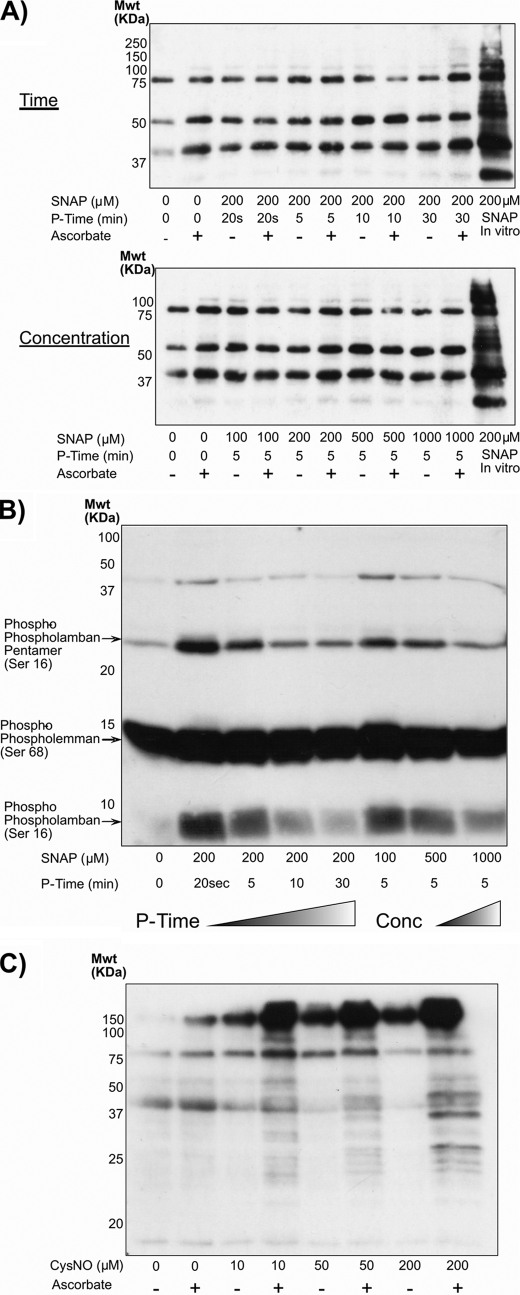
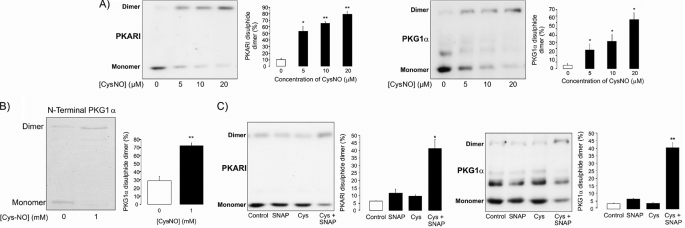
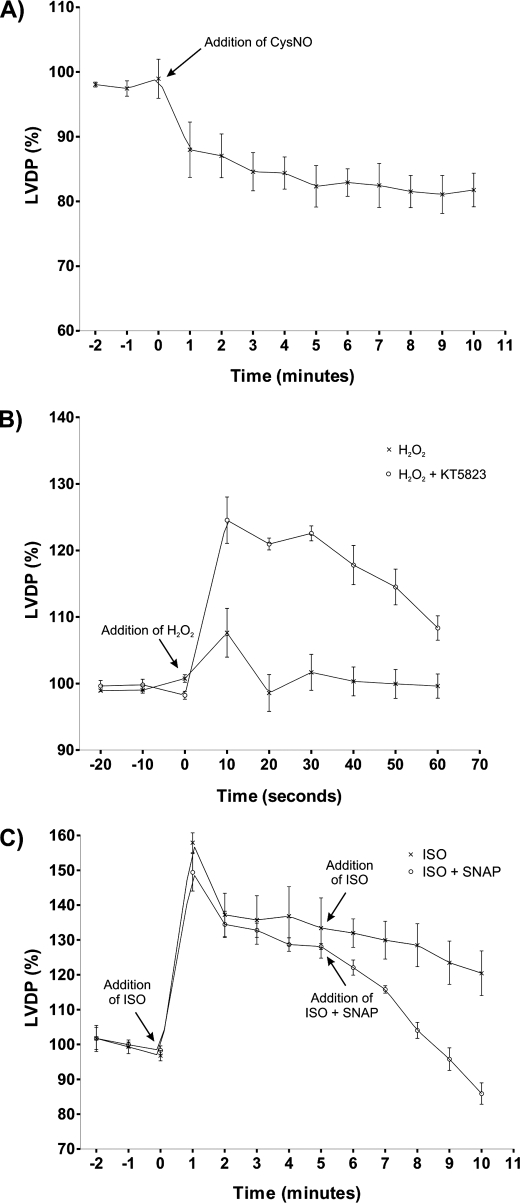
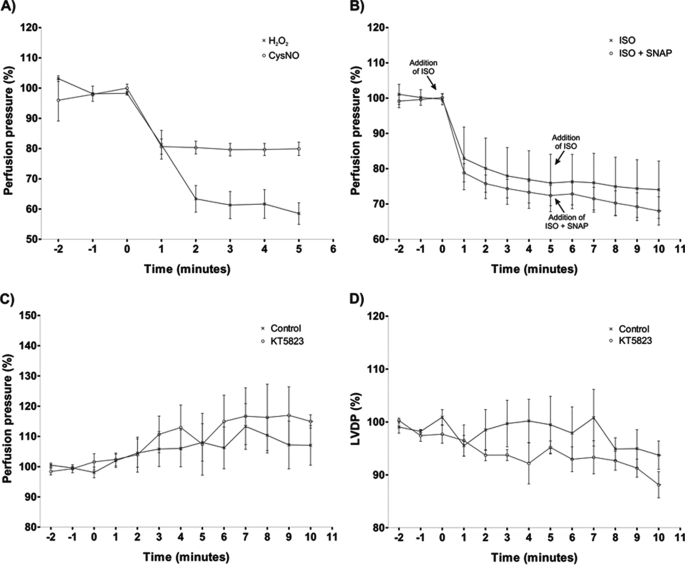
Similar articles
-
Differential role of S-nitrosylation and the NO-cGMP-PKG pathway in cardiac contractility.Nitric Oxide. 2008 May;18(3):157-67. doi: 10.1016/j.niox.2007.09.086. Epub 2007 Oct 1. Nitric Oxide. 2008. PMID: 18023373
-
Cysteine redox sensor in PKGIa enables oxidant-induced activation.Science. 2007 Sep 7;317(5843):1393-7. doi: 10.1126/science.1144318. Epub 2007 Aug 23. Science. 2007. PMID: 17717153
-
Nitric oxide regulates AKT phosphorylation and nuclear translocation in cultured retinal cells.Cell Signal. 2013 Dec;25(12):2424-39. doi: 10.1016/j.cellsig.2013.08.001. Epub 2013 Aug 17. Cell Signal. 2013. PMID: 23958999
-
Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells.Circ Res. 2003 Sep 5;93(5):406-13. doi: 10.1161/01.RES.0000091074.33584.F0. Epub 2003 Aug 14. Circ Res. 2003. PMID: 12919948
-
Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels.Comp Biochem Physiol A Mol Integr Physiol. 2005 Oct;142(2):136-43. doi: 10.1016/j.cbpb.2005.04.012. Epub 2005 May 31. Comp Biochem Physiol A Mol Integr Physiol. 2005. PMID: 15927494 Review.
Cited by
-
Obligatory role of neuronal nitric oxide synthase in the heart's antioxidant adaptation with exercise.J Mol Cell Cardiol. 2015 Apr;81:54-61. doi: 10.1016/j.yjmcc.2015.01.003. Epub 2015 Jan 14. J Mol Cell Cardiol. 2015. PMID: 25595735 Free PMC article.
-
Redox Regulation, Rather than Stress-Induced Phosphorylation, of a Hog1 Mitogen-Activated Protein Kinase Modulates Its Nitrosative-Stress-Specific Outputs.mBio. 2018 Mar 27;9(2):e02229-17. doi: 10.1128/mBio.02229-17. mBio. 2018. PMID: 29588408 Free PMC article.
-
Oxidation of Protein Kinase A Regulatory Subunit PKARIα Protects Against Myocardial Ischemia-Reperfusion Injury by Inhibiting Lysosomal-Triggered Calcium Release.Circulation. 2021 Feb 2;143(5):449-465. doi: 10.1161/CIRCULATIONAHA.120.046761. Epub 2020 Nov 13. Circulation. 2021. PMID: 33185461 Free PMC article.
-
Deficient angiogenesis in redox-dead Cys17Ser PKARIα knock-in mice.Nat Commun. 2015 Aug 10;6:7920. doi: 10.1038/ncomms8920. Nat Commun. 2015. PMID: 26258640
-
Blood Pressure-Lowering by the Antioxidant Resveratrol Is Counterintuitively Mediated by Oxidation of cGMP-Dependent Protein Kinase.Circulation. 2019 Jul 9;140(2):126-137. doi: 10.1161/CIRCULATIONAHA.118.037398. Epub 2019 May 22. Circulation. 2019. PMID: 31116951 Free PMC article.
References
-
- Stamler J. S., Lamas S., Fang F. C. (2001) Cell 106, 675–683 - PubMed
-
- Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Nat. Rev. Mol. Cell Biol. 6, 150–166 - PubMed
-
- Griffith O. W., Stuehr D. J. (1995) Annu. Rev. Physiol. 57, 707–736 - PubMed
-
- Hess D. T., Matsumoto A., Nudelman R., Stamler J. S. (2001) Nat. Cell Biol. 3, E46–E49 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
