

Large-scale analysis of Arabidopsis transcription reveals a basal co-regulation network
- PMID: 19728874
- PMCID: PMC2944327
- DOI: 10.1186/1752-0509-3-86
Large-scale analysis of Arabidopsis transcription reveals a basal co-regulation network
Abstract
Background: Analyses of gene expression data from microarray experiments has become a central tool for identifying co-regulated, functional gene modules. A crucial aspect of such analysis is the integration of data from different experiments and different laboratories. How to weigh the contribution of different experiments is an important point influencing the final outcomes. We have developed a novel method for this integration, and applied it to genome-wide data from multiple Arabidopsis microarray experiments performed under a variety of experimental conditions. The goal of this study is to identify functional globally co-regulated gene modules in the Arabidopsis genome.
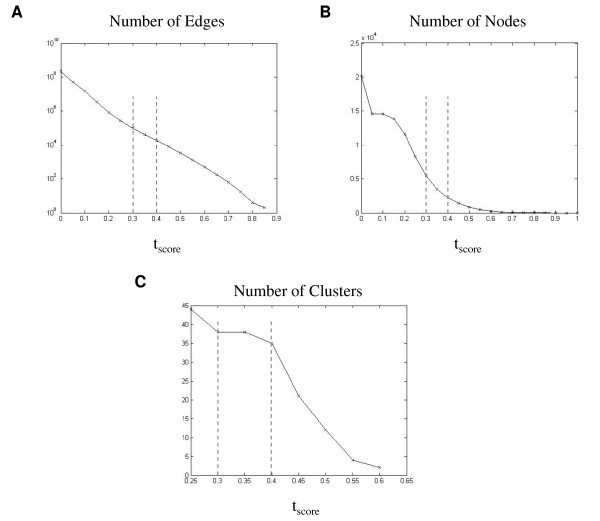
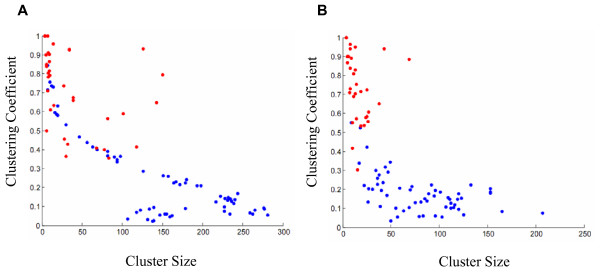
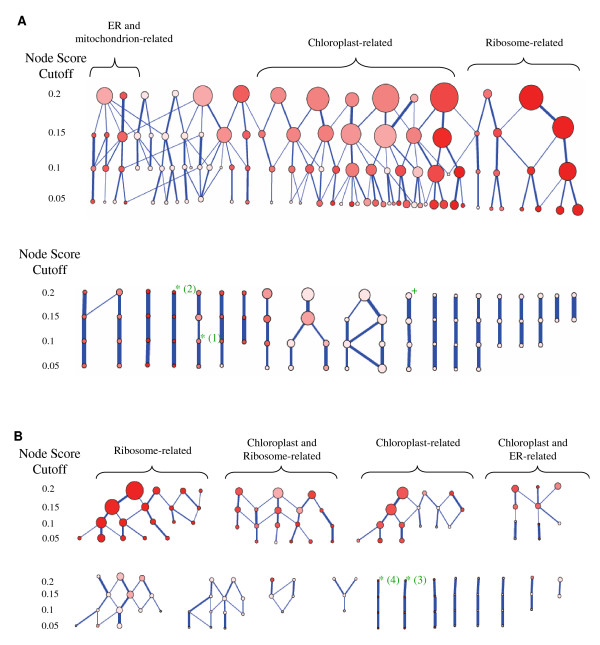
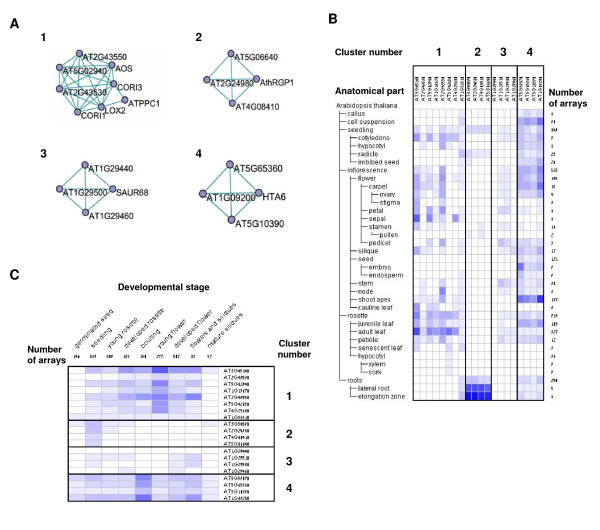
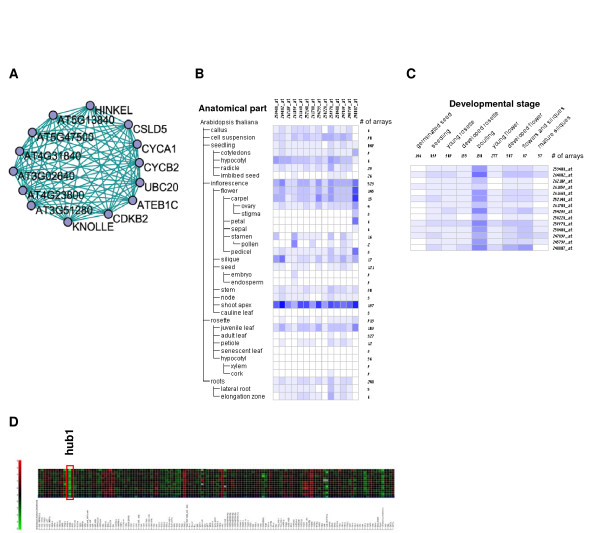
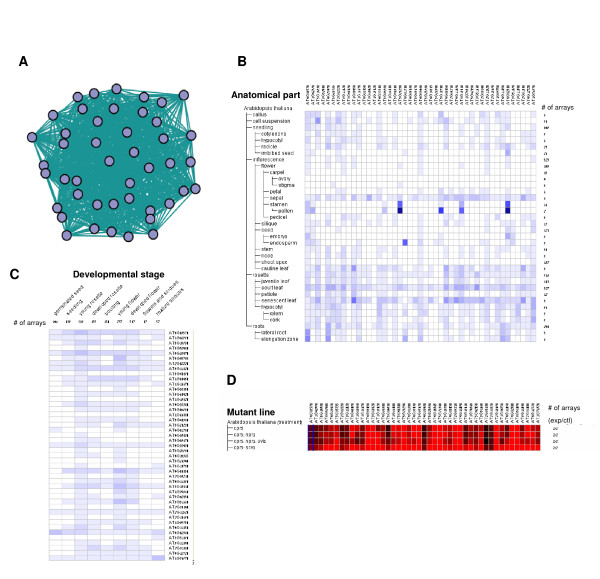
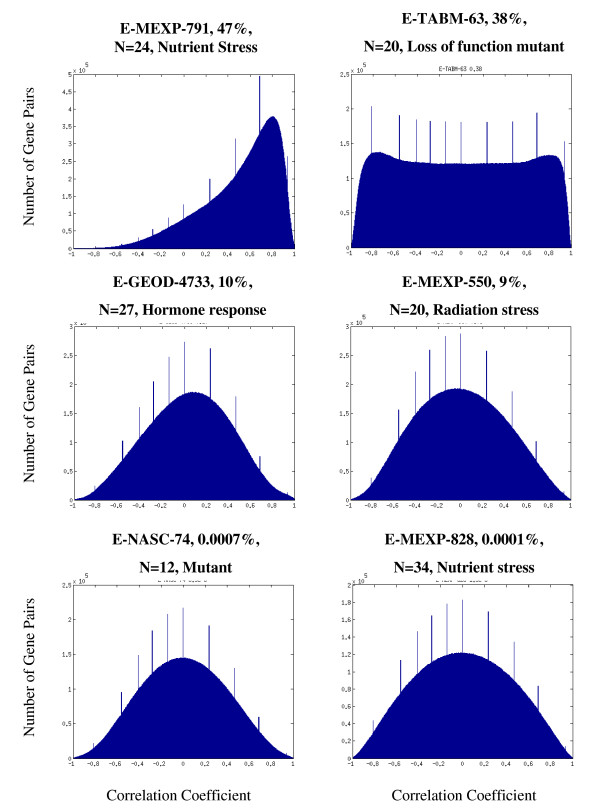
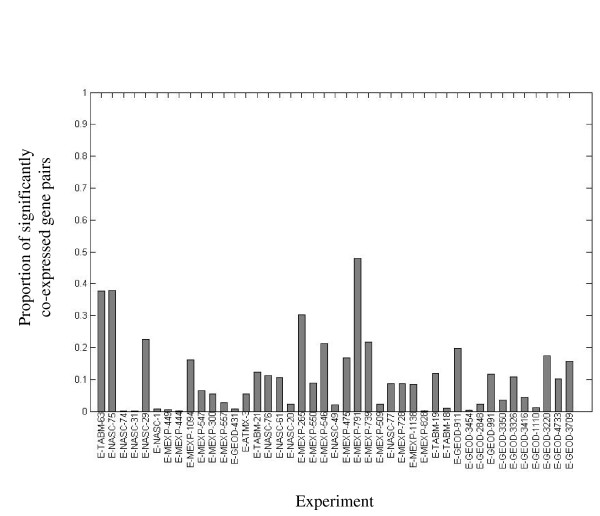
Results: Following the analysis of 21,000 Arabidopsis genes in 43 datasets and about 2 x 10(8) gene pairs, we identified a globally co-expressed gene network. We found clusters of globally co-expressed Arabidopsis genes that are enriched for known Gene Ontology annotations. Two types of modules were identified in the regulatory network that differed in their sensitivity to the node-scoring parameter; we further showed these two pertain to general and specialized modules. Some of these modules were further investigated using the Genevestigator compendium of microarray experiments. Analyses of smaller subsets of data lead to the identification of condition-specific modules.
Conclusion: Our method for identification of gene clusters allows the integration of diverse microarray experiments from many sources. The analysis reveals that part of the Arabidopsis transcriptome is globally co-expressed, and can be further divided into known as well as novel functional gene modules. Our methodology is general enough to apply to any set of microarray experiments, using any scoring function.
Figures

References
-
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci USA. 1999;96(6):2907–2912. doi: 10.1073/pnas.96.6.2907. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
