

Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells
- PMID: 19729834
- PMCID: PMC2752065
- DOI: 10.1172/JCI37978
Cyclin I activates Cdk5 and regulates expression of Bcl-2 and Bcl-XL in postmitotic mouse cells
Abstract
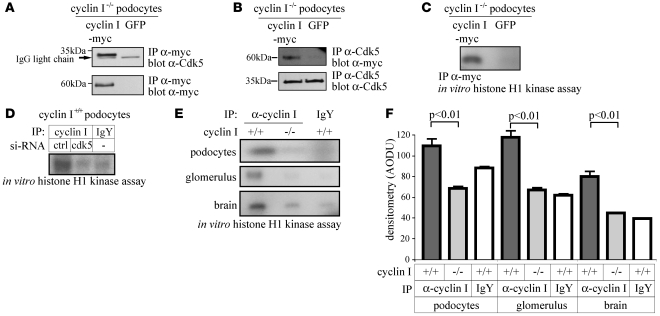
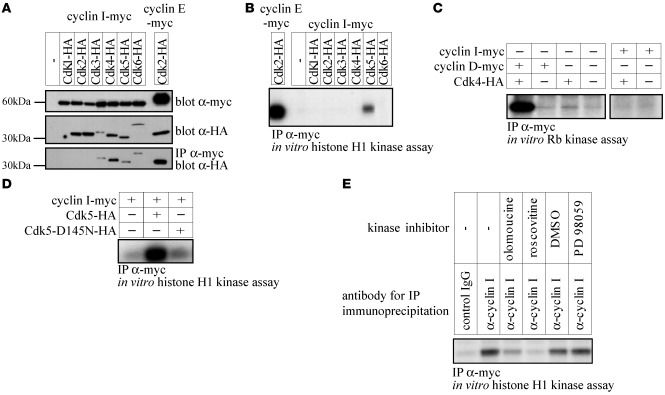
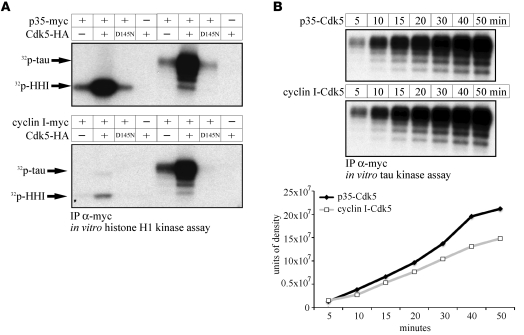
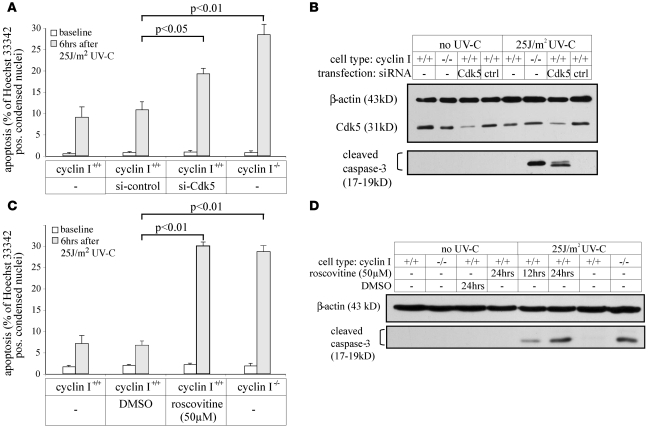
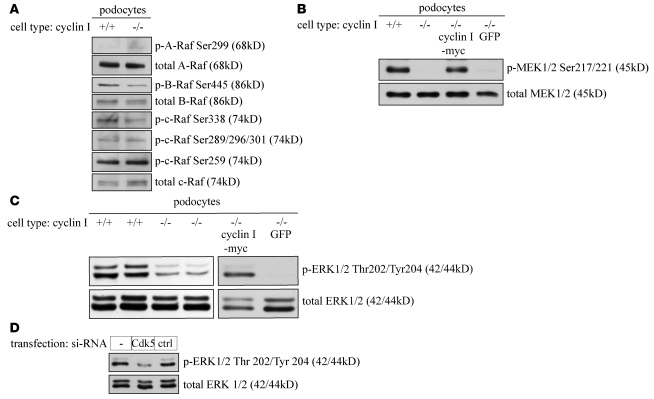
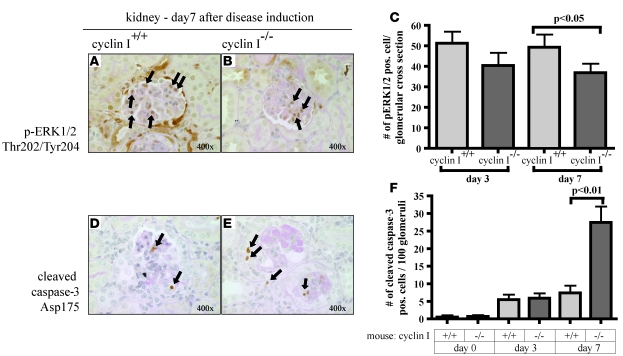
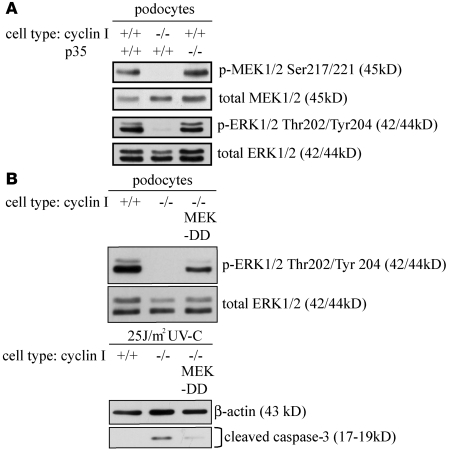
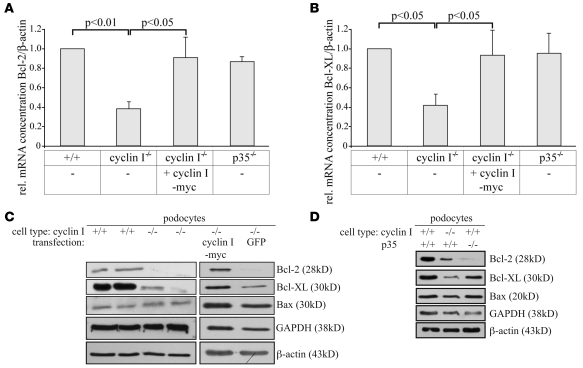
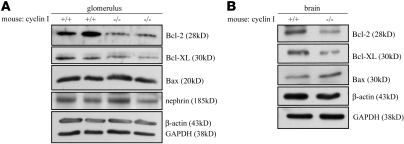
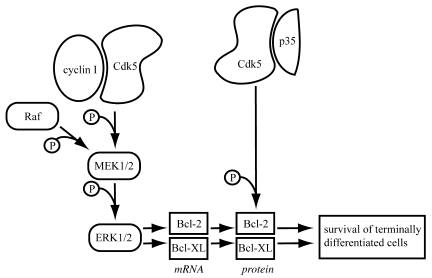
Cyclin I is an atypical cyclin because it is most abundant in postmitotic cells. We previously showed that cyclin I does not regulate proliferation, but rather controls survival of podocytes, terminally differentiated epithelial cells that are essential for the structural and functional integrity of kidney glomeruli. Here, we investigated the mechanism by which cyclin I safeguards against apoptosis and found that cyclin I bound and activated cyclin-dependent kinase 5 (Cdk5) in isolated mouse podocytes and neurons. Cdk5 activity was reduced in glomeruli and brain lysates from cyclin I-deficient mice, and inhibition of Cdk5 increased in vitro the susceptibility to apoptosis in response to cellular damage. In addition, levels of the prosurvival proteins Bcl-2 and Bcl-XL were reduced in podocytes and neurons from cyclin I-deficient mice, and restoration of Bcl-2 or Bcl-XL expression prevented injury-induced apoptosis. Furthermore, we found that levels of phosphorylated MEK1/2 and ERK1/2 were decreased in cyclin I-deficient podocytes and that inhibition of MEK1/2 restored Bcl2 and Bcl-XL protein levels. Of interest, this pathway was also defective in mice with experimental glomerulonephritis. Taken together, these data suggest that a cyclin I-Cdk5 complex forms a critical antiapoptotic factor in terminally differentiated cells that functions via MAPK signaling to modulate levels of the prosurvival proteins Bcl-2 and Bcl-XL.
Figures


References
-
- Barisoni L., Kriz W., Mundel P., D’Agati V. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 1999;10:51–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
