

The other 90% of the protein: assessment beyond the Calphas for CASP8 template-based and high-accuracy models
- PMID: 19731372
- PMCID: PMC2877634
- DOI: 10.1002/prot.22551
The other 90% of the protein: assessment beyond the Calphas for CASP8 template-based and high-accuracy models
Abstract
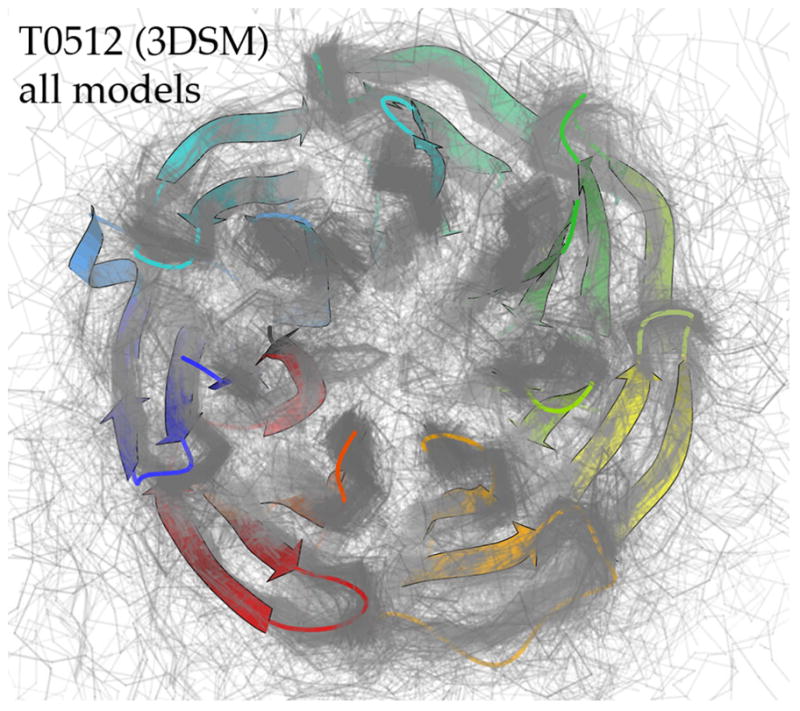
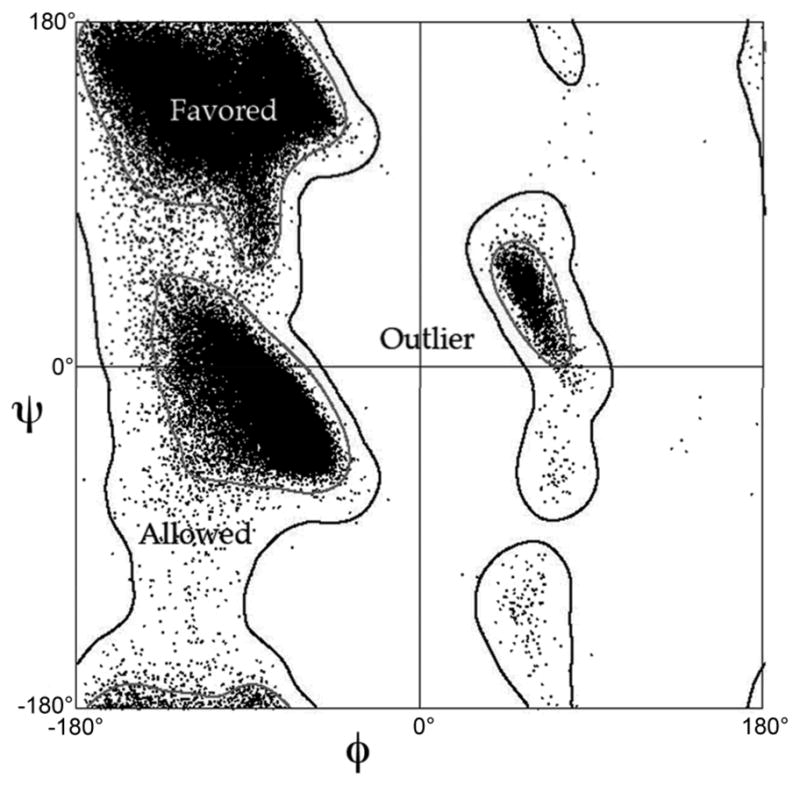
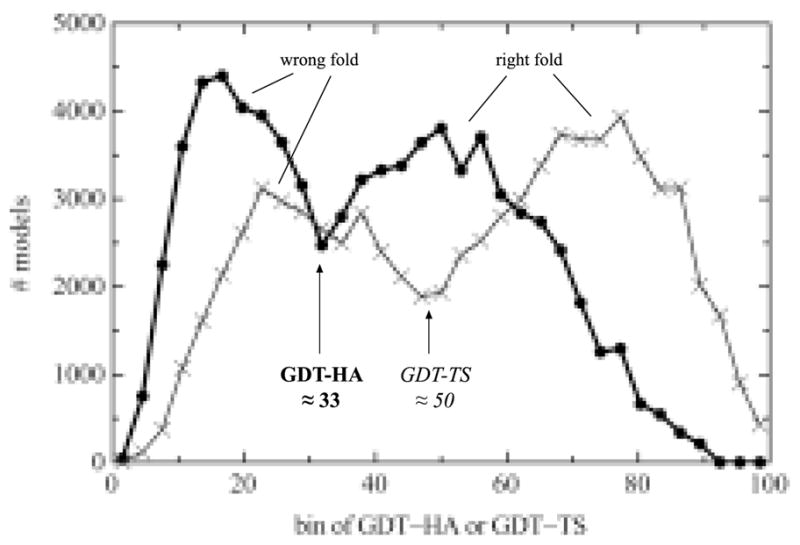
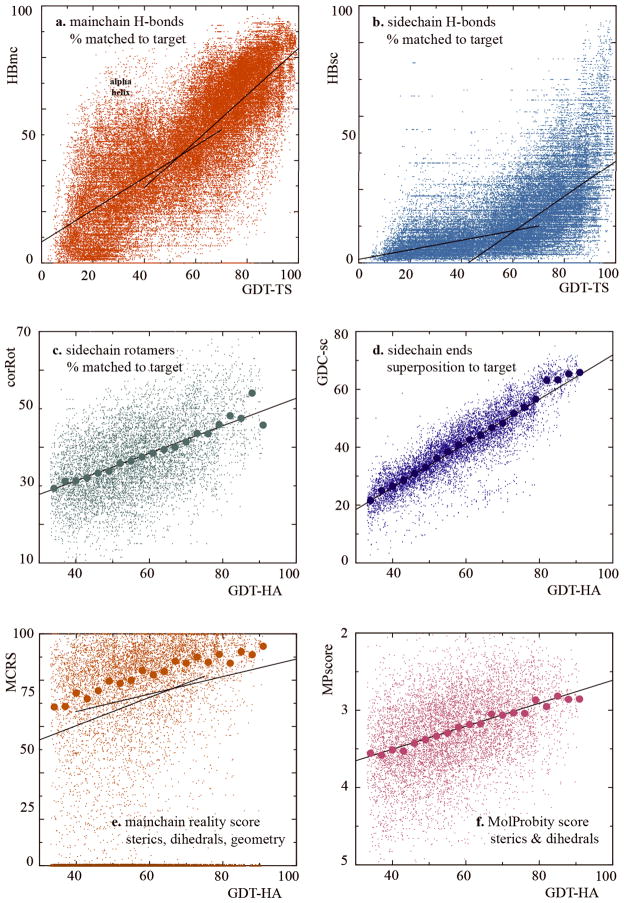
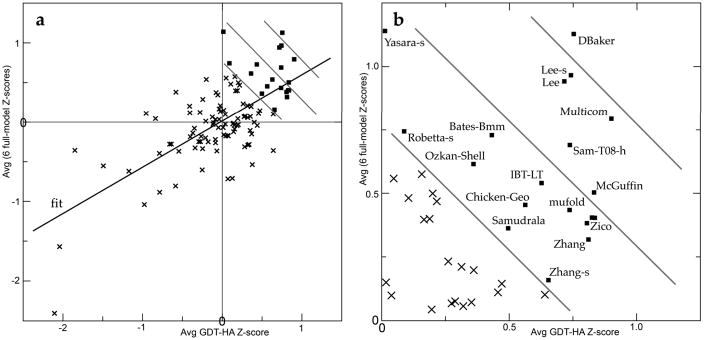
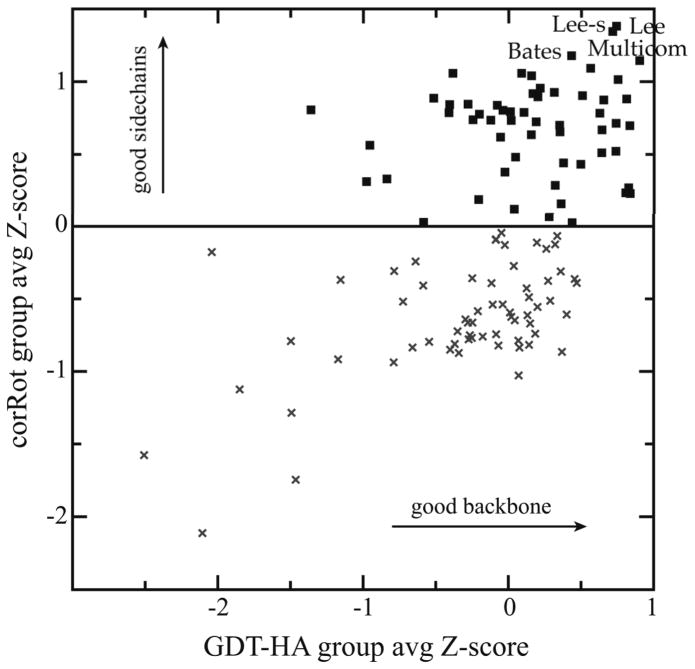
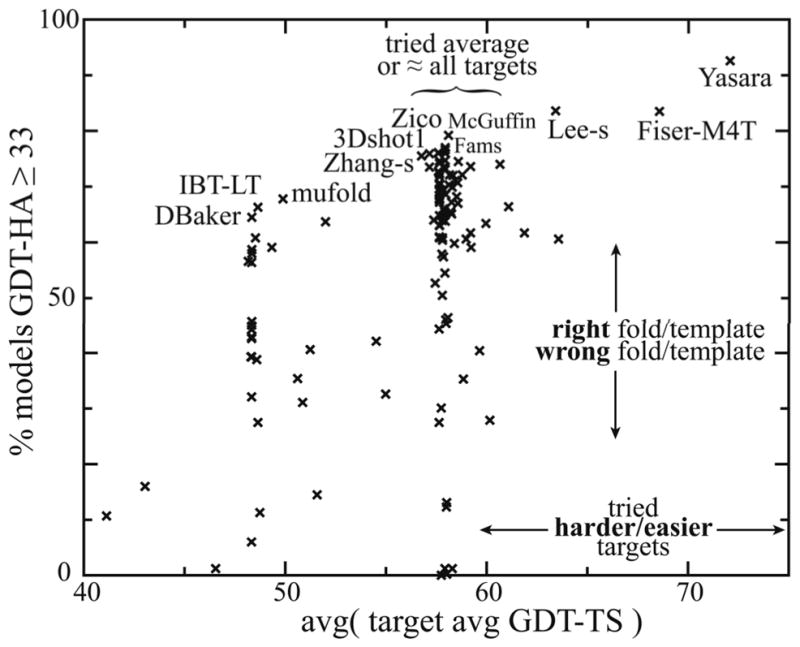
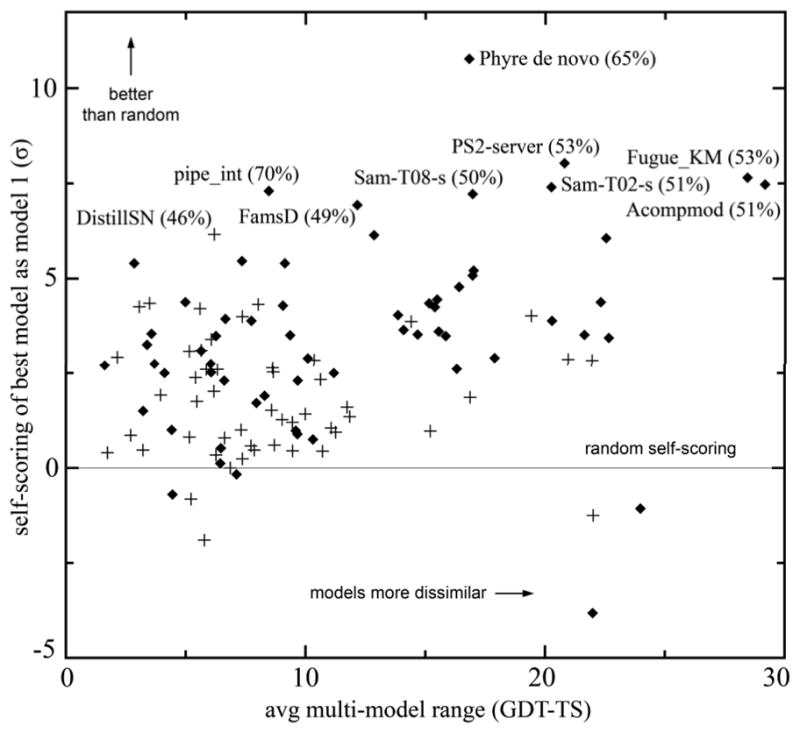
For template-based modeling in the CASP8 Critical Assessment of Techniques for Protein Structure Prediction, this work develops and applies six new full-model metrics. They are designed to complement and add value to the traditional template-based assessment by the global distance test (GDT) and related scores (based on multiple superpositions of Calpha atoms between target structure and predictions labeled "Model 1"). The new metrics evaluate each predictor group on each target, using all atoms of their best model with above-average GDT. Two metrics evaluate how "protein-like" the predicted model is: the MolProbity score used for validating experimental structures, and a mainchain reality score using all-atom steric clashes, bond length and angle outliers, and backbone dihedrals. Four other new metrics evaluate match of model to target for mainchain and sidechain hydrogen bonds, sidechain end positioning, and sidechain rotamers. Group-average Z-score across the six full-model measures is averaged with group-average GDT Z-score to produce the overall ranking for full-model, high-accuracy performance. Separate assessments are reported for specific aspects of predictor-group performance, such as robustness of approximately correct template or fold identification, and self-scoring ability at identifying the best of their models. Fold identification is distinct from but correlated with group-average GDT Z-score if target difficulty is taken into account, whereas self-scoring is done best by servers and is uncorrelated with GDT performance. Outstanding individual models on specific targets are identified and discussed. Predictor groups excelled at different aspects, highlighting the diversity of current methodologies. However, good full-model scores correlate robustly with high Calpha accuracy.
Copyright 2009 Wiley-Liss, Inc.
Figures
References
-
- Kryshtafovych A, Fidelis K, Moult J. Progress from CASP6 to CASP7. Proteins. 2007;69 (Suppl 8):194–207. - PubMed
-
- Kinch LN, Wrabl JO, Krishna SS, Majumdar I, Sadreyev RI, Qi Y, Pei J, Cheng H, Grishin NV. CASP5 assessment of fold recognition target predictions. Proteins. 2003;53 (Suppl 6):395–409. - PubMed
-
- Tress M, Ezkurdia I, Grana O, Lopez G, Valencia A. Assessment of predictions submitted for the CASP6 comparative modeling category. Proteins. 2005;61 (Suppl 7):27–45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
