

Computational analysis of membrane proteins: the largest class of drug targets
- PMID: 19733256
- PMCID: PMC2796609
- DOI: 10.1016/j.drudis.2009.08.006
Computational analysis of membrane proteins: the largest class of drug targets
Abstract
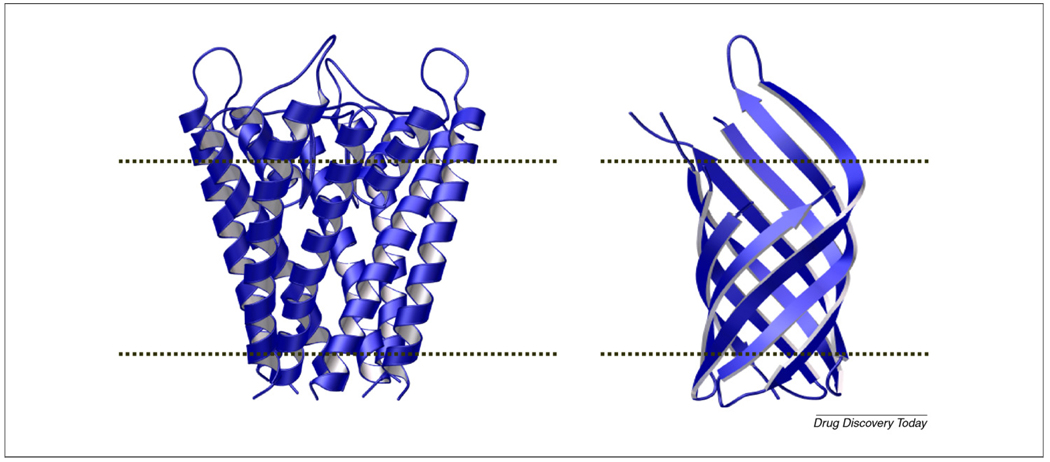
Given the key roles of integral membrane proteins as transporters and channels, it is necessary to understand their structures and, hence, mechanisms and regulation at the molecular level. Membrane proteins represent approximately 30% of all proteins of currently sequenced genomes. Paradoxically, however, only approximately 2% of crystal structures deposited in the protein data bank are of membrane proteins, and very few of these are at high resolution (better than 2A). The great disparity between our understanding of soluble proteins and our understanding of membrane proteins is because of the practical problems of working with membrane proteins - specifically, difficulties in expression, purification and crystallization. Thus, computational modeling has been utilized extensively to make crucial advances in understanding membrane protein structure and function.
Figures

References
-
- Terstappen GC, Reggiani A. In silico research in drug discovery. Trends Pharmacol. Sci. 2001;22:23–26. - PubMed
-
- Davey J. G-protein-coupled receptors: new approaches to maximise the impact of GPCRS in drug discovery. Expert Opin. Ther. Targets. 2004;8:165–170. - PubMed
-
- Xia Y, et al. Integrated prediction of the helical membrane protein interactome in yeast. J. Mol. Biol. 2006;357:339–349. - PubMed
-
- Lundstrom K. Structural genomics of GPCRs. Trends Biotechnol. 2005;23:103–108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
