

Automated inference of molecular mechanisms of disease from amino acid substitutions
- PMID: 19734154
- PMCID: PMC3140805
- DOI: 10.1093/bioinformatics/btp528
Automated inference of molecular mechanisms of disease from amino acid substitutions
Abstract
Motivation: Advances in high-throughput genotyping and next generation sequencing have generated a vast amount of human genetic variation data. Single nucleotide substitutions within protein coding regions are of particular importance owing to their potential to give rise to amino acid substitutions that affect protein structure and function which may ultimately lead to a disease state. Over the last decade, a number of computational methods have been developed to predict whether such amino acid substitutions result in an altered phenotype. Although these methods are useful in practice, and accurate for their intended purpose, they are not well suited for providing probabilistic estimates of the underlying disease mechanism.
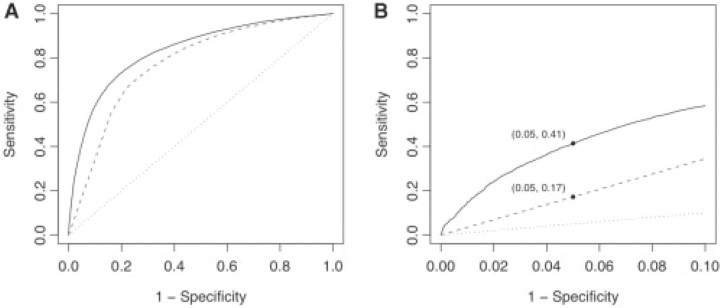
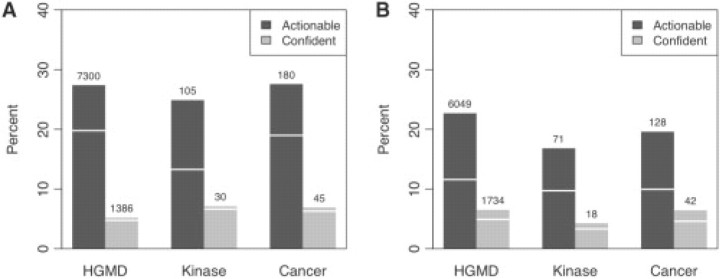
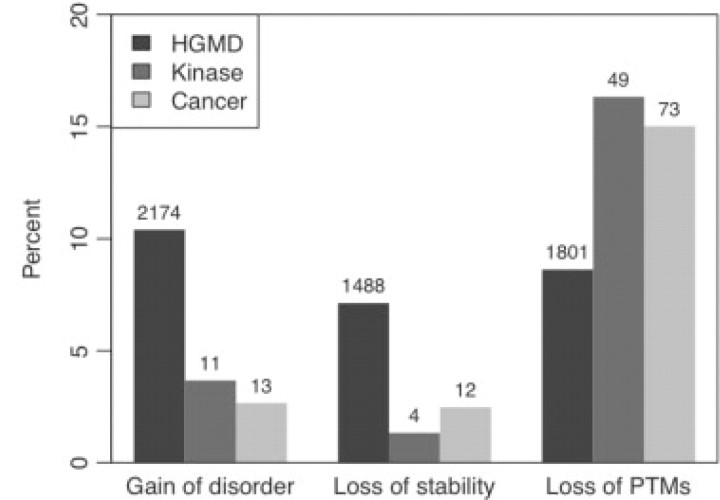
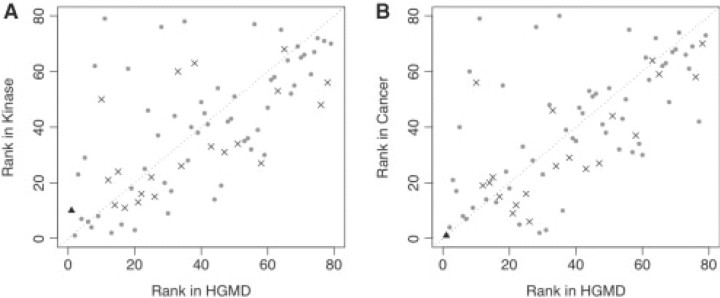
Results: We have developed a new computational model, MutPred, that is based upon protein sequence, and which models changes of structural features and functional sites between wild-type and mutant sequences. These changes, expressed as probabilities of gain or loss of structure and function, can provide insight into the specific molecular mechanism responsible for the disease state. MutPred also builds on the established SIFT method but offers improved classification accuracy with respect to human disease mutations. Given conservative thresholds on the predicted disruption of molecular function, we propose that MutPred can generate accurate and reliable hypotheses on the molecular basis of disease for approximately 11% of known inherited disease-causing mutations. We also note that the proportion of changes of functionally relevant residues in the sets of cancer-associated somatic mutations is higher than for the inherited lesions in the Human Gene Mutation Database which are instead predicted to be characterized by disruptions of protein structure.
Availability: http://mutdb.org/mutpred
Contact: predrag@indiana.edu; smooney@buckinstitute.org.
Figures
References
-
- Ahmad S, et al. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. - PubMed
-
- Bao L, Cui Y. Prediction of the phenotypic effects of non-synonymous single nucleotide polymorphisms using structural and evolutionary information. Bioinformatics. 2005;21:2185–2190. - PubMed
-
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
