

Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross
- PMID: 19734204
- PMCID: PMC2888688
- DOI: 10.1101/sqb.2009.74.013
Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross
Abstract
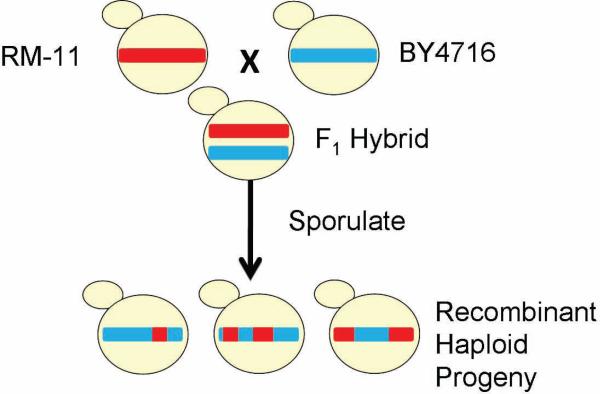
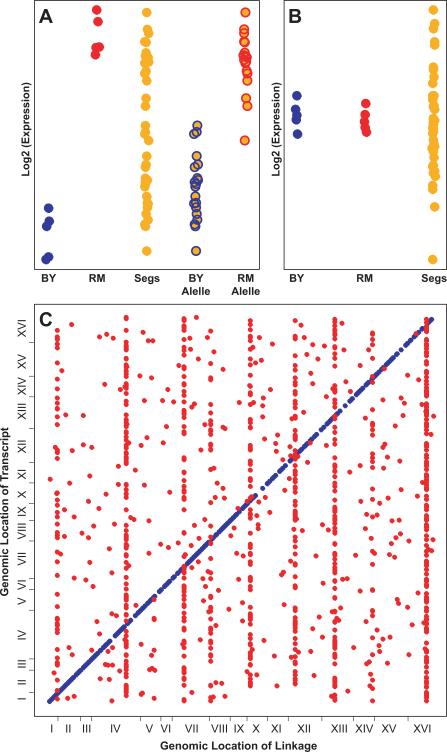
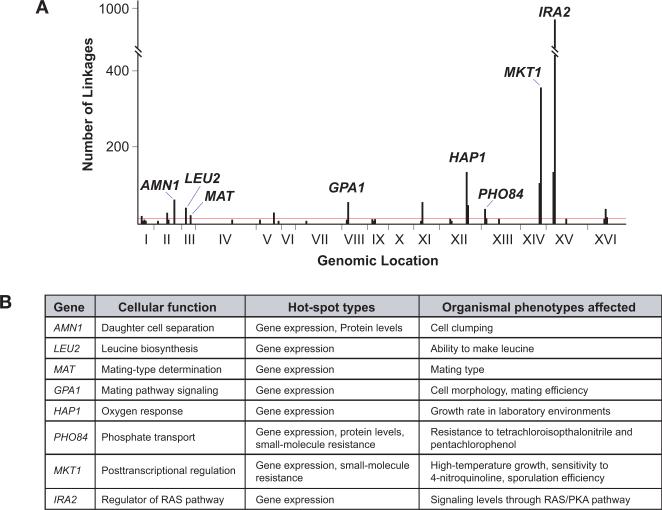
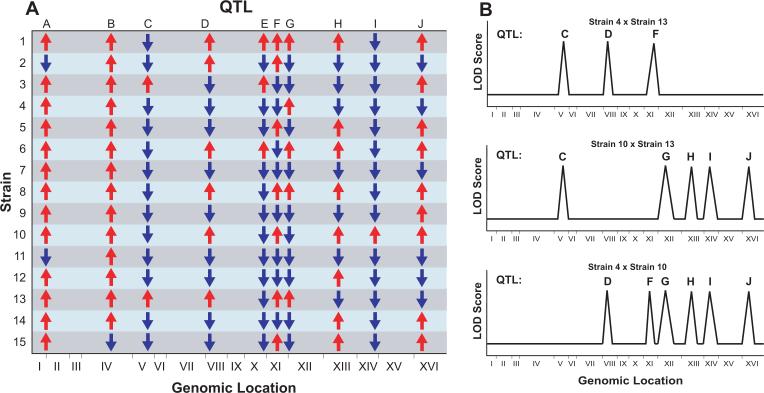
The genetic basis of many phenotypes of biological and medical interest, including susceptibility to common human diseases, is complex, involving multiple genes that interact with one another and the environment. Despite decades of effort, we possess neither a full grasp of the general rules that govern complex trait genetics nor a detailed understanding of the genetic basis of specific complex traits. We have used a cross between two yeast strains, BY and RM, to systematically investigate the genetic complexity underlying differences in global gene expression and other traits. The number and diversity of traits dissected to the locus, gene, and nucleotide levels in the BYxRM cross make it arguably the most extensively characterized system with regard to causal effects of genetic variation on phenotype. We summarize the insights obtained to date into the genetics of complex traits in yeast, with an emphasis on the BYxRM cross. We then highlight the central outstanding questions about the genetics of complex traits and discuss how to answer them using yeast as a model system.
Figures
Similar articles
-
High-resolution mapping of complex traits with a four-parent advanced intercross yeast population.Genetics. 2013 Nov;195(3):1141-55. doi: 10.1534/genetics.113.155515. Epub 2013 Sep 13. Genetics. 2013. PMID: 24037264 Free PMC article.
-
Genetic architecture of highly complex chemical resistance traits across four yeast strains.PLoS Genet. 2012;8(3):e1002570. doi: 10.1371/journal.pgen.1002570. Epub 2012 Mar 15. PLoS Genet. 2012. PMID: 22438822 Free PMC article.
-
Finding the sources of missing heritability in a yeast cross.Nature. 2013 Feb 14;494(7436):234-7. doi: 10.1038/nature11867. Epub 2013 Feb 3. Nature. 2013. PMID: 23376951 Free PMC article.
-
Advances in quantitative trait analysis in yeast.PLoS Genet. 2012;8(8):e1002912. doi: 10.1371/journal.pgen.1002912. Epub 2012 Aug 16. PLoS Genet. 2012. PMID: 22916041 Free PMC article. Review.
-
The molecular basis of phenotypic variation in yeast.Curr Opin Genet Dev. 2013 Dec;23(6):672-7. doi: 10.1016/j.gde.2013.10.005. Epub 2013 Nov 21. Curr Opin Genet Dev. 2013. PMID: 24269094 Free PMC article. Review.
Cited by
-
History and Domestication of Saccharomyces cerevisiae in Bread Baking.Front Genet. 2020 Nov 11;11:584718. doi: 10.3389/fgene.2020.584718. eCollection 2020. Front Genet. 2020. PMID: 33262788 Free PMC article. Review.
-
Local ancestry corrects for population structure in Saccharomyces cerevisiae genome-wide association studies.Genetics. 2012 Dec;192(4):1503-11. doi: 10.1534/genetics.112.144790. Epub 2012 Sep 28. Genetics. 2012. PMID: 23023004 Free PMC article.
-
Germplasm Resources and Strategy for Genetic Breeding of Lycium Species: A Review.Front Plant Sci. 2022 Feb 11;13:802936. doi: 10.3389/fpls.2022.802936. eCollection 2022. Front Plant Sci. 2022. PMID: 35222468 Free PMC article. Review.
-
Saccharomyces cerevisiae FLO1 Gene Demonstrates Genetic Linkage to Increased Fermentation Rate at Low Temperatures.G3 (Bethesda). 2017 Mar 10;7(3):1039-1048. doi: 10.1534/g3.116.037630. G3 (Bethesda). 2017. PMID: 28143947 Free PMC article.
-
A widespread inversion polymorphism conserved among Saccharomyces species is caused by recurrent homogenization of a sporulation gene family.PLoS Genet. 2022 Nov 28;18(11):e1010525. doi: 10.1371/journal.pgen.1010525. eCollection 2022 Nov. PLoS Genet. 2022. PMID: 36441813 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases