

Peroxisome proliferator-activated receptor-gamma ligands induce heme oxygenase-1 in lung fibroblasts by a PPARgamma-independent, glutathione-dependent mechanism
- PMID: 19734319
- PMCID: PMC2777492
- DOI: 10.1152/ajplung.00148.2009
Peroxisome proliferator-activated receptor-gamma ligands induce heme oxygenase-1 in lung fibroblasts by a PPARgamma-independent, glutathione-dependent mechanism
Abstract
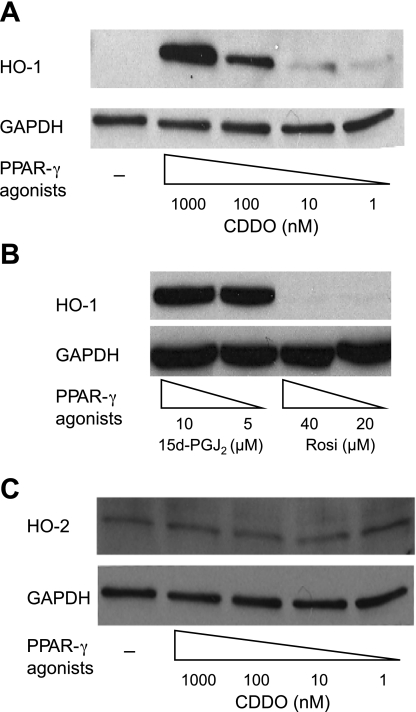
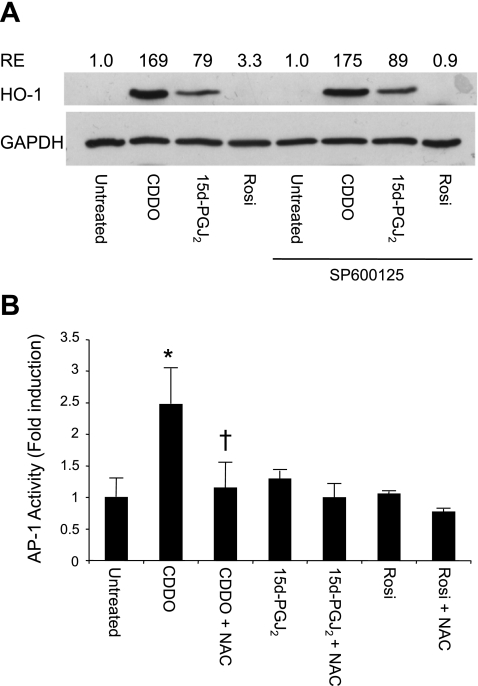
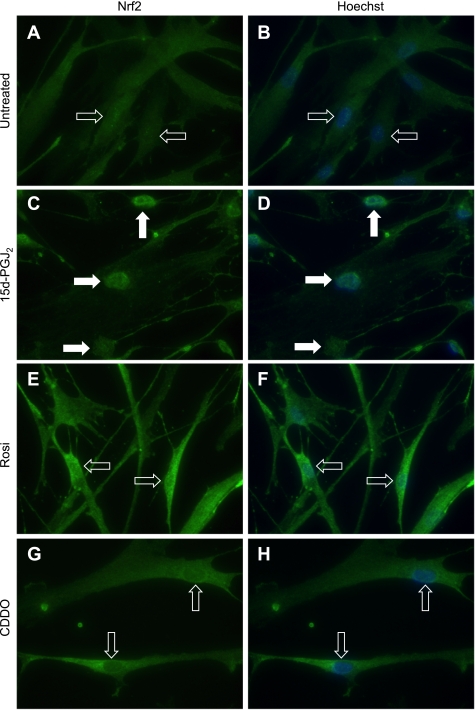
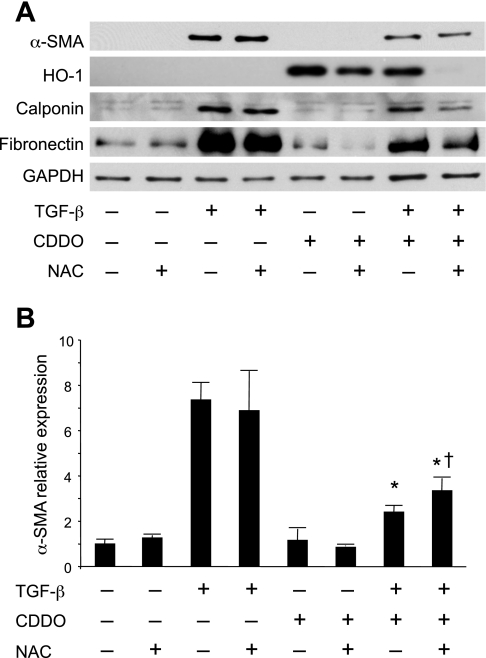
Oxidative stress plays an important role in the pathogenesis of pulmonary fibrosis. Heme oxygenase-1 (HO-1) is a key antioxidant enzyme, and overexpression of HO-1 significantly decreases lung inflammation and fibrosis in animal models. Peroxisome proliferator-activated receptor-gamma (PPARgamma) is a transcription factor that regulates adipogenesis, insulin sensitization, and inflammation. We report here that the PPARgamma ligands 15d-PGJ2 and 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid (CDDO), which have potent antifibrotic effects in vitro, also strongly induce HO-1 expression in primary human lung fibroblasts. Pharmacological and genetic approaches are used to demonstrate that induction of HO-1 is PPARgamma independent. Upregulation of HO-1 coincides with decreased intracellular glutathione (GSH) levels and can be inhibited by N-acetyl cysteine (NAC), a thiol antioxidant and GSH precursor. Upregulation of HO-1 is not inhibited by Trolox, a non-thiol antioxidant, and does not involve the transcription factors AP-1 or Nrf2. CDDO and 15d-PGJ2 contain an alpha/beta unsaturated ketone that acts as an electrophilic center that can form covalent bonds with free reduced thiols. Rosiglitazone, a PPARgamma ligand that lacks an electrophilic center, does not induce HO-1. These data suggest that in human lung fibroblasts, 15d-PGJ2 and CDDO induce HO-1 via a GSH-dependent mechanism involving the formation of covalent bonds between 15d-PGJ2 or CDDO and GSH. Inhibiting HO-1 upregulation with NAC has only a small effect on the antifibrotic properties of 15d-PGJ2 and CDDO in vitro. These results suggest that CDDO and similar electrophilic PPARgamma ligands may have great clinical potential as antifibrotic agents, not only through direct effects on fibroblast differentiation and function, but indirectly by bolstering antioxidant defenses.
Figures


Similar articles
-
Harnessing peroxisome proliferator-activated receptor γ agonists to induce Heme Oxygenase-1: a promising approach for pulmonary inflammatory disorders.Cell Commun Signal. 2024 Feb 15;22(1):125. doi: 10.1186/s12964-024-01501-4. Cell Commun Signal. 2024. PMID: 38360670 Free PMC article. Review.
-
Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules.Am J Respir Cell Mol Biol. 2014 Apr;50(4):737-47. doi: 10.1165/rcmb.2013-0092OC. Am J Respir Cell Mol Biol. 2014. PMID: 24175906 Free PMC article.
-
Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts.Am J Respir Cell Mol Biol. 2009 Dec;41(6):722-30. doi: 10.1165/rcmb.2009-0006OC. Epub 2009 Mar 13. Am J Respir Cell Mol Biol. 2009. PMID: 19286977 Free PMC article.
-
Electrophilic PPARγ ligands inhibit corneal fibroblast to myofibroblast differentiation in vitro: a potentially novel therapy for corneal scarring.Exp Eye Res. 2012 Jan;94(1):136-45. doi: 10.1016/j.exer.2011.11.018. Epub 2011 Dec 8. Exp Eye Res. 2012. PMID: 22178289 Free PMC article.
-
Peroxisome proliferator-activated receptor-gamma (PPARγ) and its immunomodulation function: current understanding and future therapeutic implications.Expert Rev Clin Pharmacol. 2022 Mar;15(3):295-303. doi: 10.1080/17512433.2022.2071697. Epub 2022 May 1. Expert Rev Clin Pharmacol. 2022. PMID: 35481412 Review.
Cited by
-
Harnessing peroxisome proliferator-activated receptor γ agonists to induce Heme Oxygenase-1: a promising approach for pulmonary inflammatory disorders.Cell Commun Signal. 2024 Feb 15;22(1):125. doi: 10.1186/s12964-024-01501-4. Cell Commun Signal. 2024. PMID: 38360670 Free PMC article. Review.
-
Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules.Am J Respir Cell Mol Biol. 2014 Apr;50(4):737-47. doi: 10.1165/rcmb.2013-0092OC. Am J Respir Cell Mol Biol. 2014. PMID: 24175906 Free PMC article.
-
Normal Human Lung Epithelial Cells Inhibit Transforming Growth Factor-β Induced Myofibroblast Differentiation via Prostaglandin E2.PLoS One. 2015 Aug 6;10(8):e0135266. doi: 10.1371/journal.pone.0135266. eCollection 2015. PLoS One. 2015. PMID: 26248335 Free PMC article.
-
PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO.PPAR Res. 2012;2012:641087. doi: 10.1155/2012/641087. Epub 2012 Mar 5. PPAR Res. 2012. PMID: 22481913 Free PMC article.
-
Metabolic dysregulation in pulmonary fibrosis: insights into amino acid contributions and therapeutic potential.Cell Death Discov. 2025 Aug 27;11(1):411. doi: 10.1038/s41420-025-02715-2. Cell Death Discov. 2025. PMID: 40858556 Free PMC article. Review.
References
-
- Antonicelli F, Brown D, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Regulation of LPS-mediated inflammation in vivo and in vitro by the thiol antioxidant Nacystelyn. Am J Physiol Lung Cell Mol Physiol 286: L1319–L1327, 2004 - PubMed
-
- Baglole CJ, Bushinsky SM, Garcia TM, Kode A, Rahman I, Sime PJ, Phipps RP. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 291: L19–L29, 2006 - PubMed
-
- Baglole CJ, Reddy SY, Pollock SJ, Feldon SE, Sime PJ, Smith TJ, Phipps RP. Isolation and phenotypic characterization of lung fibroblasts. Methods Mol Med 117: 115–127, 2005 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources