

Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression
- PMID: 19734907
- PMCID: PMC2756031
- DOI: 10.1038/nmeth.1370
Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression
Abstract
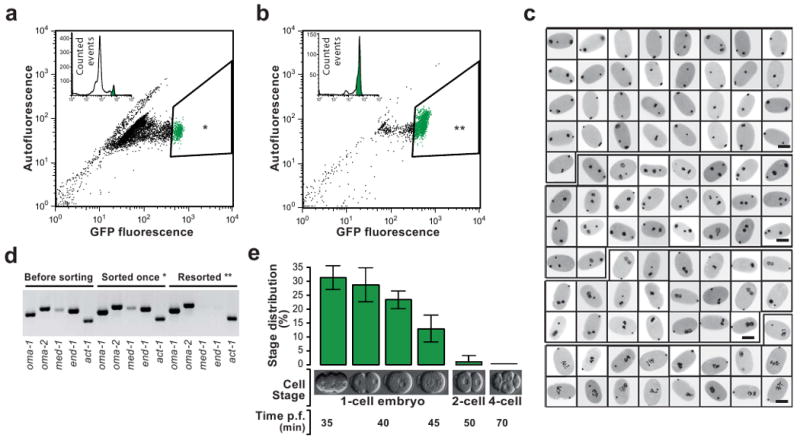
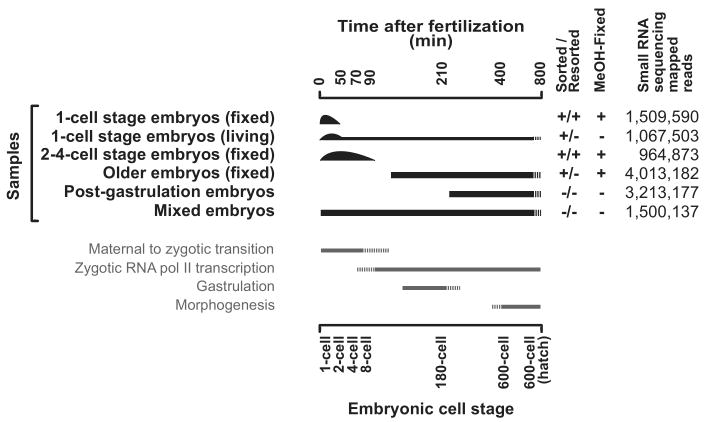
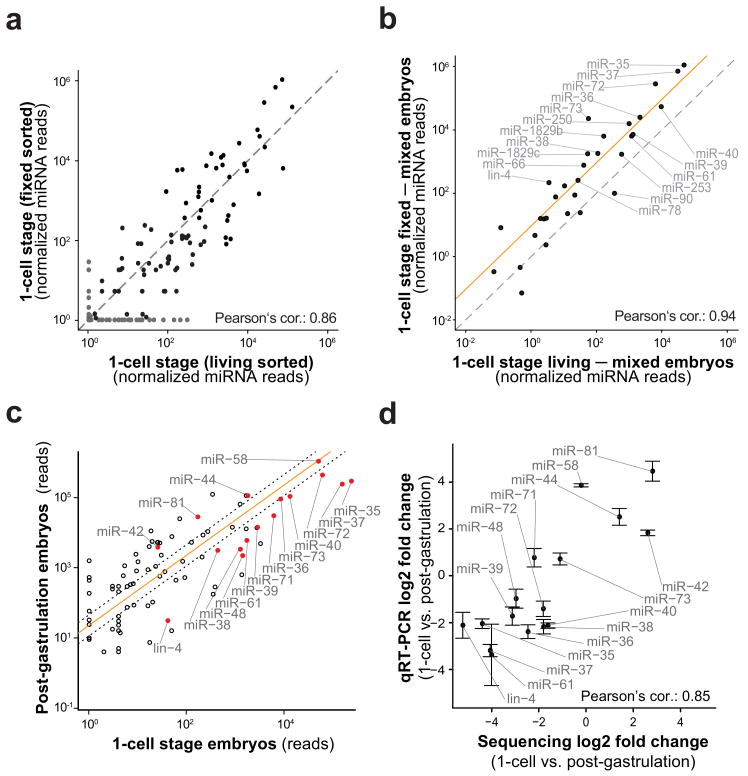
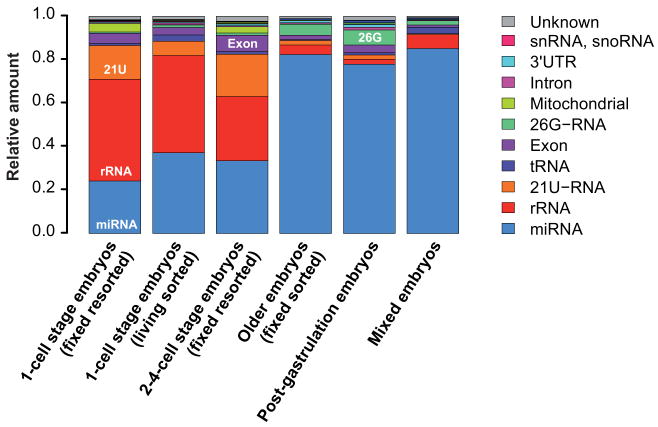
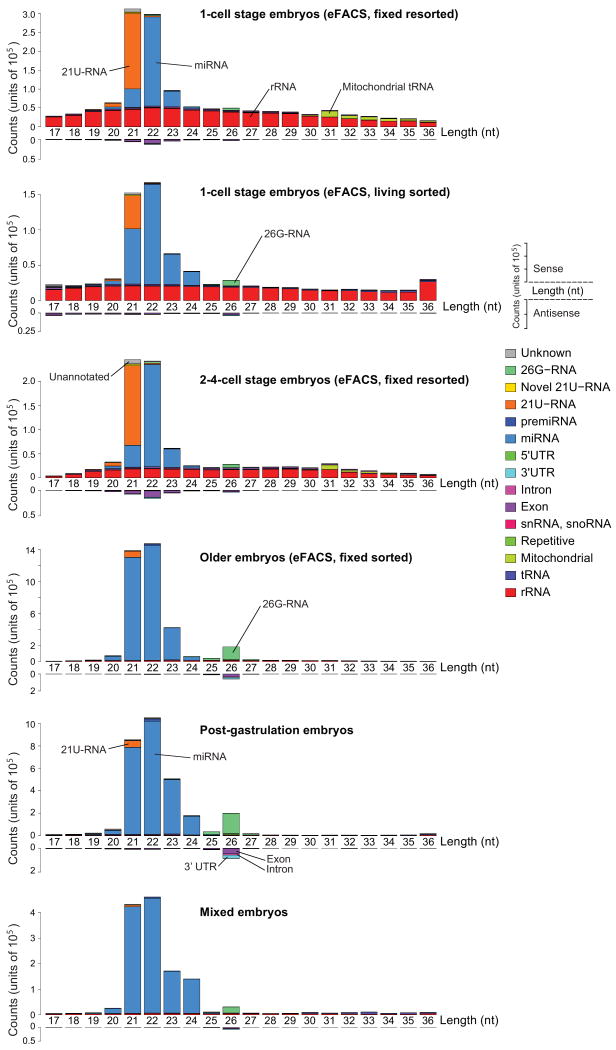
Caenorhabditis elegans is one of the most prominent model systems for embryogenesis, but collecting many precisely staged embryos has been impractical. Thus, early C. elegans embryogenesis has not been amenable to most high-throughput genomics or biochemistry assays. To overcome this problem, we devised a method to collect staged C. elegans embryos by fluorescence-activated cell sorting (eFACS). In a proof-of-principle experiment, we found that a single eFACS run routinely yielded tens of thousands of almost perfectly staged 1-cell stage embryos. As the earliest embryonic events are driven by posttranscriptional regulation, we combined eFACS with second-generation sequencing to profile the embryonic expression of small, noncoding RNAs. We discovered complex and orchestrated changes in the expression between and within almost all classes of small RNAs, including microRNAs and 26G-RNAs, during embryogenesis.
Figures

Comment in
-
Automated sorting of live C. elegans using laFACS.Nat Methods. 2010 Jun;7(6):417-8. doi: 10.1038/nmeth.f.304. Epub 2010 May 2. Nat Methods. 2010. PMID: 20436474 Free PMC article. No abstract available.
References
-
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. - PubMed
-
- Piano F, et al. RNAi analysis of genes expressed in the ovary of Caenorhabditis elegans. Curr Biol. 2000;10:1619–1622. - PubMed
-
- Piano F, et al. Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr Biol. 2002;12:1959–1964. - PubMed
-
- Kamath RS, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
