

Cerebral blood flow response to functional activation
- PMID: 19738630
- PMCID: PMC2872188
- DOI: 10.1038/jcbfm.2009.188
Cerebral blood flow response to functional activation
Abstract
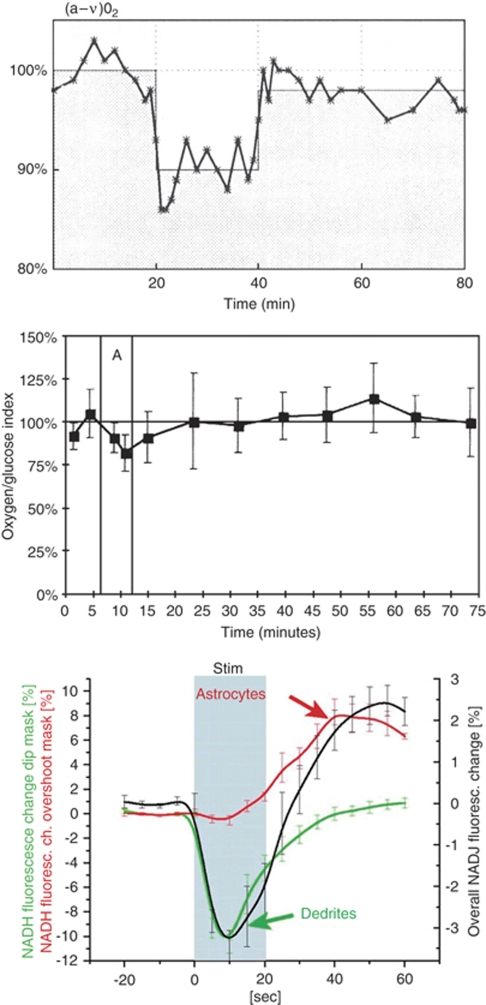
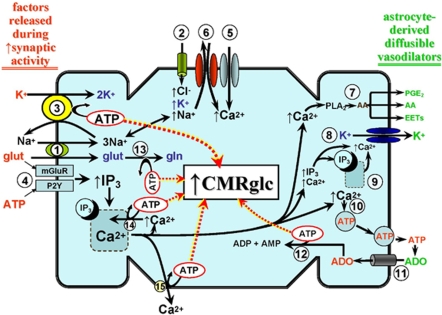
Cerebral blood flow (CBF) and cerebral metabolic rate are normally coupled, that is an increase in metabolic demand will lead to an increase in flow. However, during functional activation, CBF and glucose metabolism remain coupled as they increase in proportion, whereas oxygen metabolism only increases to a minor degree-the so-called uncoupling of CBF and oxidative metabolism. Several studies have dealt with these issues, and theories have been forwarded regarding the underlying mechanisms. Some reports have speculated about the existence of a potentially deficient oxygen supply to the tissue most distant from the capillaries, whereas other studies point to a shift toward a higher degree of non-oxidative glucose consumption during activation. In this review, we argue that the key mechanism responsible for the regional CBF (rCBF) increase during functional activation is a tight coupling between rCBF and glucose metabolism. We assert that uncoupling of rCBF and oxidative metabolism is a consequence of a less pronounced increase in oxygen consumption. On the basis of earlier studies, we take into consideration the functional recruitment of capillaries and attempt to accommodate the cerebral tissue's increased demand for glucose supply during neural activation with recent evidence supporting a key function for astrocytes in rCBF regulation.
Figures
References
-
- Ances BM, Buerk DG, Greenberg JH, Detre JA. Temporal dynamics of the partial pressure of brain tissue oxygen during functional forepaw stimulation in rats. Neurosci Lett. 2001;306:106–110. - PubMed
-
- Anderson AW, Heptulla RA, Driesen N, Flanagan D, Goldberg PA, Jones TW, Rife F, Sarofin H, Tamborlane W, Sherwin R, Gore JC. Effects of hypoglycemia on human brain activation measured with fMRI. Magn Reson Imaging. 2006;24:693–697. - PubMed
-
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. - PubMed
-
- Barros LF, Porras OH, Bittner CX. Why glucose transport in the brain matters for PET. Trends Neurosci. 2005;28:117–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
