

Review
doi: 10.1146/annurev-pathol-121808-102113.
Mutational heterogeneity in human cancers: origin and consequences
Affiliations
- PMID: 19743960
- PMCID: PMC3375045
- DOI: 10.1146/annurev-pathol-121808-102113
Item in Clipboard
Review
Mutational heterogeneity in human cancers: origin and consequences
Annu Rev Pathol.
2010.
Abstract
Cancer recapitulates Darwinian evolution. Mutations acquired during life that provide cells with a growth or survival advantage will preferentially multiply to form a tumor. As a result of The Cancer Genome Atlas Project, we have gathered detailed information on the nucleotide sequence changes in a number of human cancers. The sources of mutations in cancer are diverse, and the complexity of those found to be clonally present in tumors has increasingly made it difficult to identify key rate-limiting genes for tumor growth that could serve as potential targets for directed therapies. The impact of DNA sequencing on future cancer research and personalized therapy is likely to be profound and merits critical evaluation.
Figures
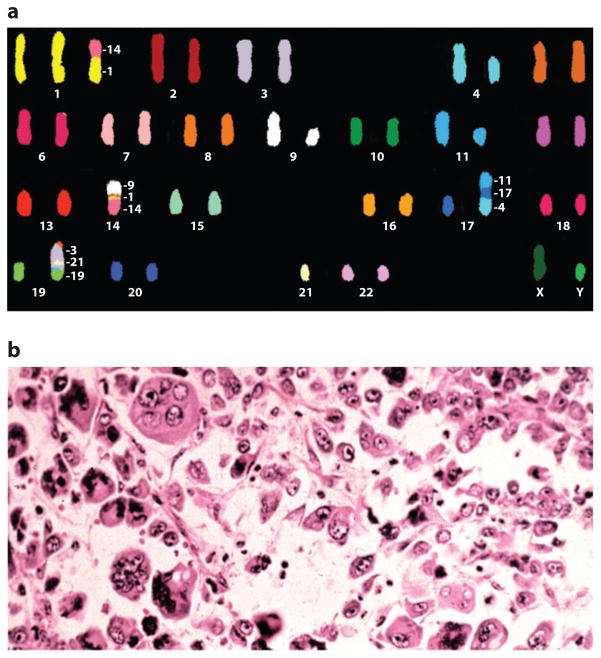
A) Chromosomal heterogeneity. Spectral karyotype from an acute myelogenous leukemia (AML) cell demonstrating aneuploidy and multiple chromosomal rearrangements. Courtesy of Dr. Karen Swisshelm, Department of Pathology, University of Colorado, Denver. B) Morphologic heterogeneity. Hematoxylin and eosin section from a large cell, undifferentiated lung cancer demonstrating a highly pleiomorphic cellular population. Courtesy of Dr. Ray Monnat, Department of Pathology, University of Washington.
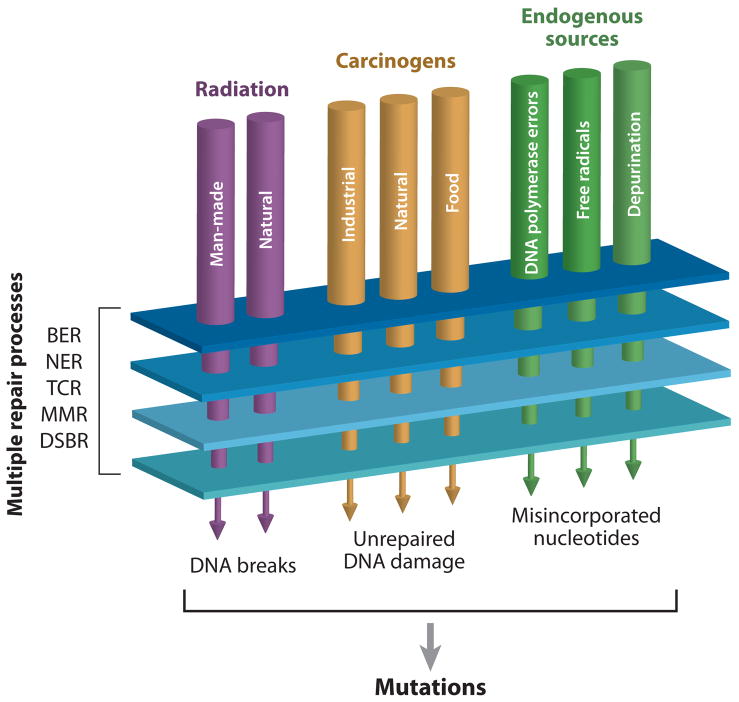
In each human cell DNA is damaged thousands of times per day by both exogenous and endogenous sources. Most alterations are corrected by cellular mechanisms including: base excision repair (BER), nucleotide excision repair (NER), transcription coupled repair (TCR), mismatch repair (MMR) and double strand break repair (DSBR). Lesions that escape repair have the potential to cause mutations during DNA replication.
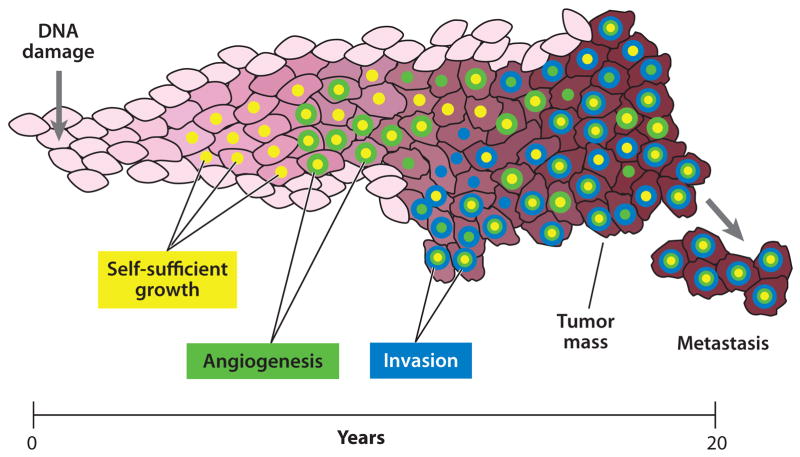
Within a developing tumor mutations accumulate over time as a result of unrepaired DNA damage. Most of these are either neutral or detrimental; only a small number bestow a cell with growth and survival benefits. These beneficial variants will preferentially multiply and produce additional mutations that may undergo further selection and expansion. Adventagous phenotypes for tumor growth include, among others, the ability to divide independently of extracellular signals (yellow), the ability to recruit a blood supply (green) and the ability to invade adjacent and distant tissues (blue). After overcoming all antineoplastic defense mechanisms, a tumor may proliferate indefinitely until the death of the host.
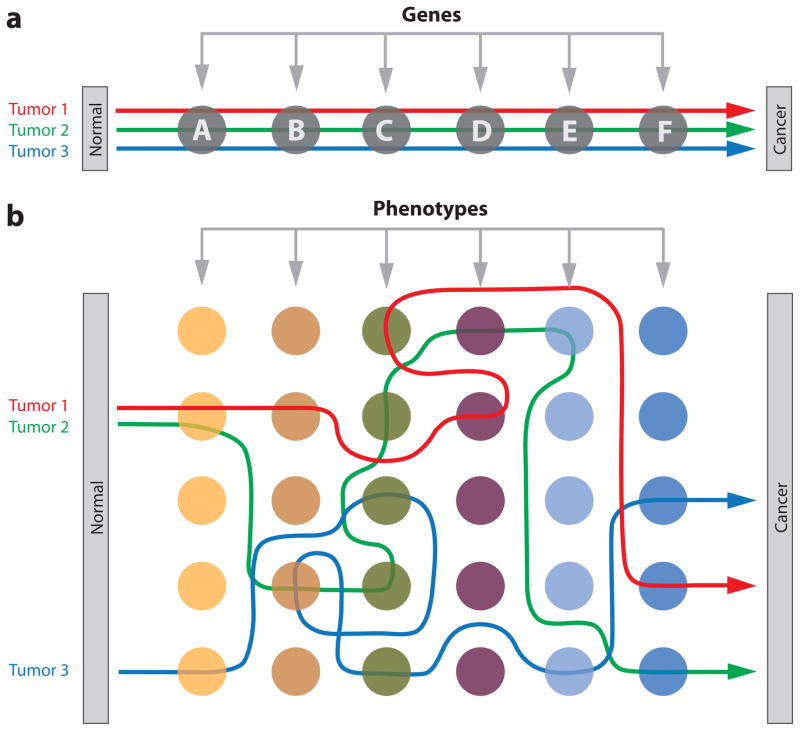
A) Deterministic. In this model, different tumors of the same cancer type occur reproducibly through sequential mutation of each gene within a defined series. Although mutation occurs randomly, the order of selection is fixed. B) Plastic. In this model, different tumors evolve along highly variable pathways, selecting for specific cancer phenotypes that may be achieved through (epi)mutation of many possible sites in the genome. Although some mutated loci may be shared by different tumors, most are not and the order of selection is predominantly stochastic.
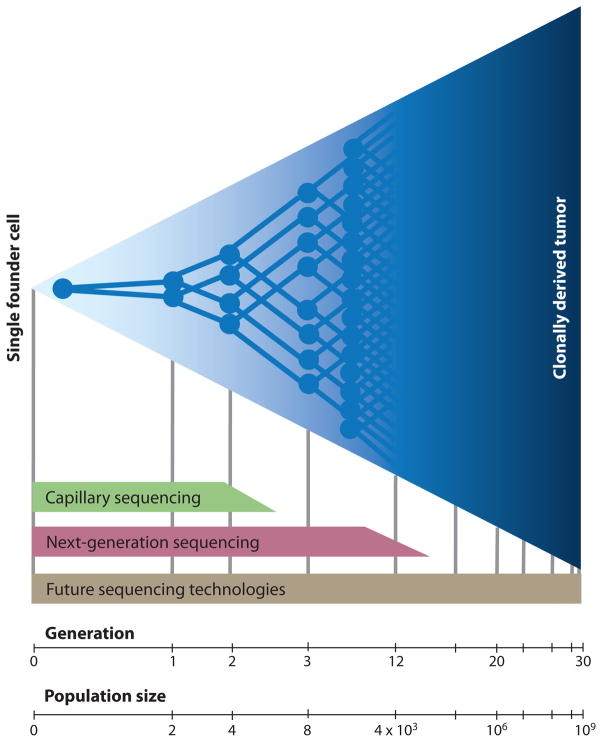
Depicted is the clonal expansion of a single cell into a population of one billion cells. In a hypothetical scenario where no cell death occurs, this requires approximately 30 generations of division. Current capillary methods of DNA sequencing only detect mutations that have clonally expanded to represent 25% or more of a tumor. Only mutations that are present in the founding cell or that arise within the first two generations of division can be identified. Deep sequencing on current “next-generation” sequencing platforms are reported to detect subclonal mutations down to a frequency of 1/5000 (119) (thus, those occurring within the first twelve generations after founding). Sensitivity is limited by the error rate of PCR amplification steps and that of the sequencing chemistry itself. Future technologies may eventually enable ultra-accurate, high-throughput detection of mutations that arise during any stage of clonal expansion.
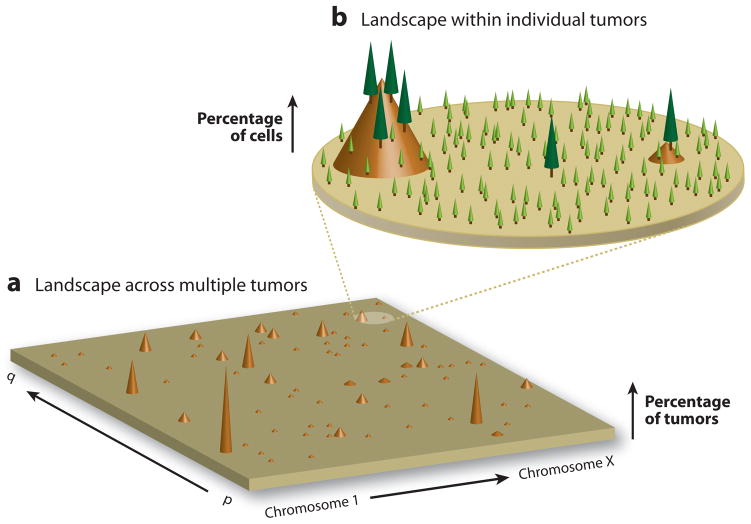
A) The cancer genome landscape proposed by Wood et. al. (88) graphically represents the mutational heterogeneity among different tumors of a single cancer type. The height of each brown peak indicates the percentage of tumors found to carry a clonal mutation in a particular gene. The landscape comprises a small number of “mountains”–genes which are clonally mutated in a large fraction of individual cancers–and a significantly greater number of “hills” – genes clonally mutated in only one or a few tumors. While there may be 50 or more genes clonally mutated within the genome of an individual tumor, most genes are rarely mutated in more than a few tumors. B) An additional level of the mutational landscape exists within individual tumors due to differences among the genomes of single tumor cells. Although a small number of mutations are clonally present in the majority of cells in an individual tumor (“trees”), an exponentially larger number exist subclonally in only one or a few cells (“seedlings”). Among this vast reservoir of non-clonal mutations exists many therapy-resistant variants.
References
-
- Boveri T. Veh Dtsch Zool Ges. Wurzburg: 1902. Uber mehrpolige Mitosen als Mittel zur Analyse des Zellkerns.
-
- Cleaver JE, Kraemer KH. Xeroderma pigmentosum. In: Scriver CR, Beudet AL, Sktm WS, Valle D, editors. Metabolic Basis of Inherited Disease. New York, NY: McGraw-Hill; 1989. pp. 2949–71. Number of 2949–71 pp.
-
- Riopel MA, Spellerberg A, Griffin CA, Perlman EJ. Genetic analysis of ovarian germ cell tumors by comparative genomic hybridization. Cancer Res. 1998;58:3105–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
