

Addressing reported pro-apoptotic functions of NF-kappaB: targeted inhibition of canonical NF-kappaB enhances the apoptotic effects of doxorubicin
- PMID: 19746155
- PMCID: PMC2734988
- DOI: 10.1371/journal.pone.0006992
Addressing reported pro-apoptotic functions of NF-kappaB: targeted inhibition of canonical NF-kappaB enhances the apoptotic effects of doxorubicin
Abstract
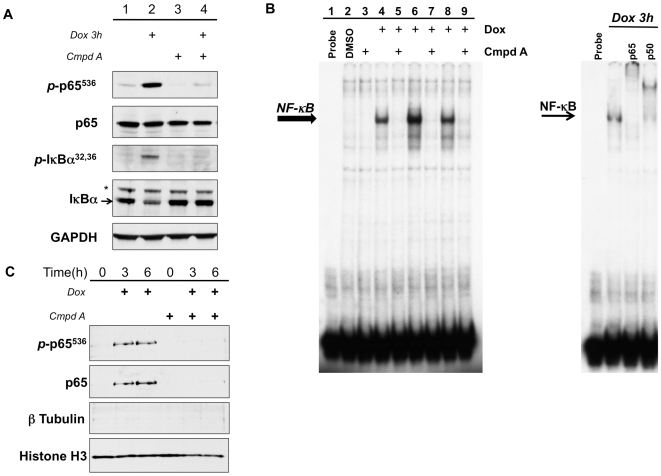
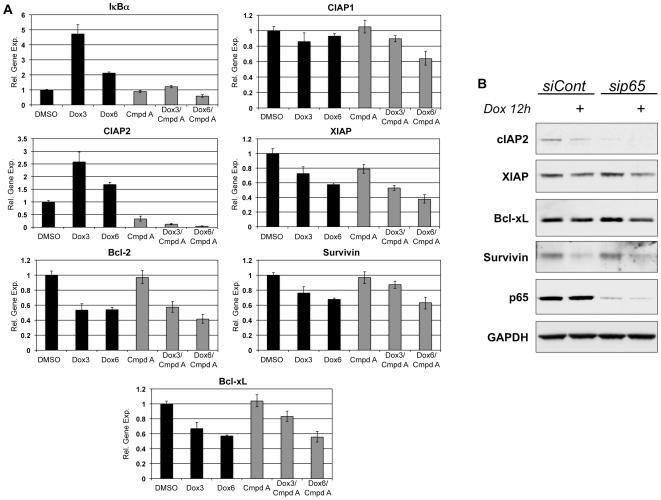
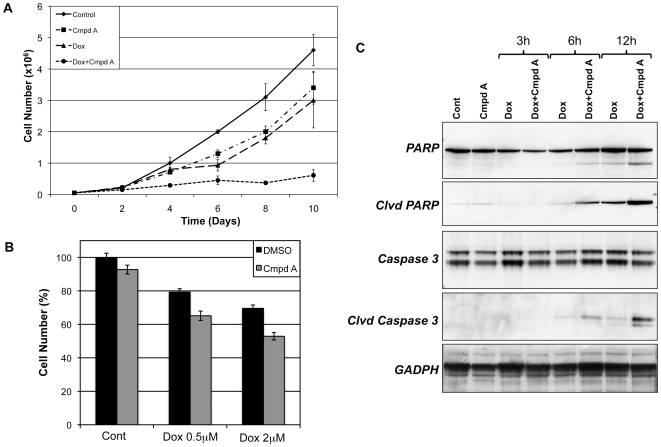
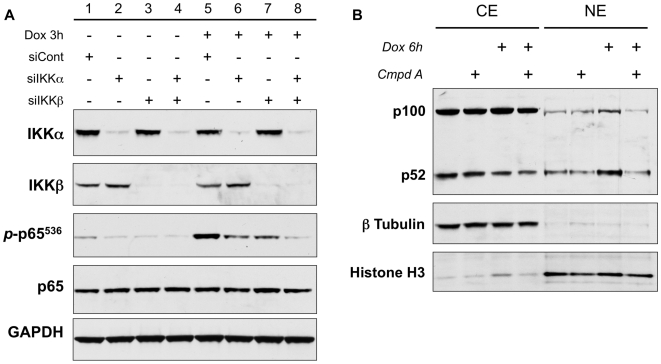
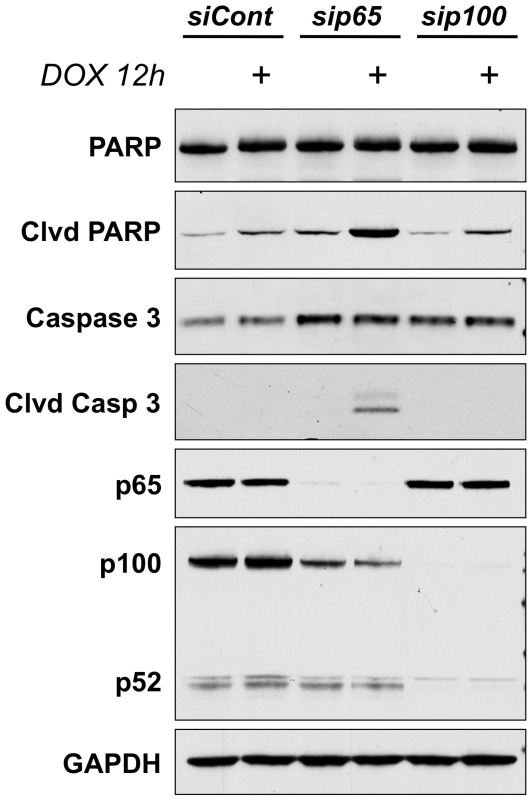
The ability of the transcription factor NF-kappaB to upregulate anti-apoptotic proteins has been linked to the chemoresistance of solid tumors to standard chemotherapy. In contrast, recent studies have proposed that, in response to doxorubicin, NF-kappaB can be pro-apoptotic through repression of anti-apoptotic target genes. However, there is little evidence analyzing the outcome of NF-kappaB inhibition on the cytotoxicity of doxorubicin in studies describing pro-apoptotic NF-kappaB activity. In this study, we further characterize the activation of NF-kappaB in response to doxorubicin and evaluate its role in chemotherapy-induced cell death in sarcoma cells where NF-kappaB is reported to be pro-apoptotic. Doxorubicin treatment in U2OS cells induced canonical NF-kappaB activity as evidenced by increased nuclear accumulation of phosphorylated p65 at serine 536 and increased DNA-binding activity. Co-treatment with a small molecule IKKbeta inhibitor, Compound A, abrogated this response. RT-PCR evaluation of anti-apoptotic gene expression revealed that doxorubicin-induced transcription of cIAP2 was inhibited by Compound A, while doxorubicin-induced repression of other anti-apoptotic genes was unaffected by Compound A or siRNA to p65. Furthermore, the combination of doxorubicin and canonical NF-kappaB inhibition with Compound A or siRNA to p65 resulted in decreased cell viability measured by trypan blue staining and MTS assay and increased apoptosis measured by cleaved poly (ADP-ribose) polymerase and cleaved caspase 3 when compared to doxorubicin alone. Our results demonstrate that doxorubicin-induced canonical NF-kappaB activity associated with phosphorylated p65 is anti-apoptotic in its function and that doxorubicin-induced repression of anti-apoptotic genes occurs independent of p65. Therefore, combination therapies incorporating NF-kappaB inhibitors together with standard chemotherapies remains a viable method to improve the clinical outcomes in patients with advanced stage malignancies.
Conflict of interest statement
Figures
References
-
- Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. - PubMed
-
- Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. - PubMed
-
- Shen HM, Tergaonkar V. NFkappaB signaling in carcinogenesis and as a potential molecular target for cancer therapy. Apoptosis. 2009;14:348–363. - PubMed
-
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
