

Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study
- PMID: 19749005
- PMCID: PMC2803118
- DOI: 10.1378/chest.09-1002
Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study
Abstract
Background: Limited data exist regarding the population-based epidemiology of idiopathic pulmonary fibrosis (IPF). The objective of the study was to describe the trends in the incidence, prevalence, and clinical course of IPF in the community.
Methods: We conducted a population-based study of adult patients with IPF in Olmsted County, Minnesota, from 1997 to 2005. Two methods were used to identify IPF cases, as defined by the 2002 American Thoracic Society/European Respiratory Society consensus statement: (1) usual interstitial pneumonia (UIP) on a surgical lung biopsy specimen or a definite UIP pattern on a high-resolution CT image (narrow criteria) and (2) UIP on a surgical lung biopsy specimen or a definite or possible UIP pattern on CT image (broad criteria).
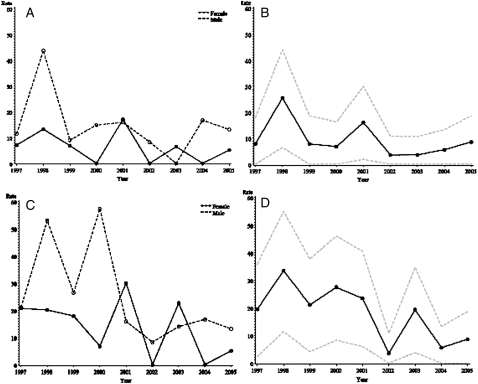
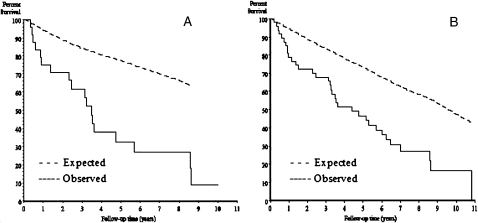
Results: Of 596 patients screened for the possibility of pulmonary disease or pulmonary fibrosis over 9 years of follow-up, 47 cases had IPF. Of these, 24 met the narrow criteria. The age- and sex-adjusted incidence was 8.8/100,000 and 17.4/100,000 person-years, for narrow and broad criteria, respectively. The age-adjusted incidence was higher in men than in women, and among patients aged 70-79 years. During the study period, the incidence of IPF decreased (P < .001). On December 31, 2005, the age- and sex-adjusted prevalence was 27.9/100,000 and 63/100,000 persons by narrow and broad criteria, respectively. Thirty-seven patients experienced a total of 53 respiratory exacerbations (26 IPF related, 27 non-IPF related), and 34 (72%) patients died. The primary cause of death was IPF related in 16 (47%) patients. Median survival for narrow-criteria and broad-criteria incidence cases was 3.5 and 4.4 years, respectively.
Conclusions: The incidence of IPF in Olmsted County decreased over the study period. Nonprimary IPF respiratory exacerbations are as frequent as primary IPF respiratory exacerbations and an important cause of death.
Figures
References
-
- Roelandt M, Demedts M, Callebaut W, et al. Epidemiology of interstitial lung disease (ILD) in Flanders: registration by pneumologists in 1992-1994. Working group on ILD, VRGT. Vereniging voor Respiratoire Gezondheidszorg en Tuberculosebestrijding. Acta Clin Belg. 1995;50(5):260–268. - PubMed
-
- Schweisfurth H, Kieslich C, Satake N, et al. How are interstitial lung diseases diagnosed in Germany? Results of the scientific registry for the exploration of interstitial lung diseases (“Fibrosis Registry”) of the WATL [in German] Pneumologie. 2003;57(7):373–382. - PubMed
-
- Thomeer M, Demedts M, Vandeurzen K VRGT Working Group on Interstitial Lung Diseases. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg. 2001;56(3):163–172. - PubMed
-
- von Plessen C, Grinde O, Gulsvik A. Incidence and prevalence of cryptogenic fibrosing alveolitis in a Norwegian community. Respir Med. 2003;97(4):428–435. - PubMed
-
- Xaubet A, Ancochea J, Morell F, et al. Spanish Group on Interstitial Lung Diseases, SEPAR. Report on the incidence of interstitial lung diseases in Spain. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(1):64–70. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
