

Fast mapping of short sequences with mismatches, insertions and deletions using index structures
- PMID: 19750212
- PMCID: PMC2730575
- DOI: 10.1371/journal.pcbi.1000502
Fast mapping of short sequences with mismatches, insertions and deletions using index structures
Abstract
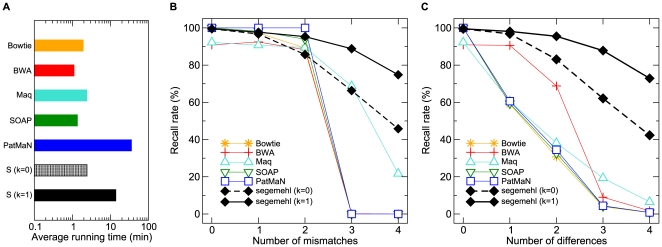
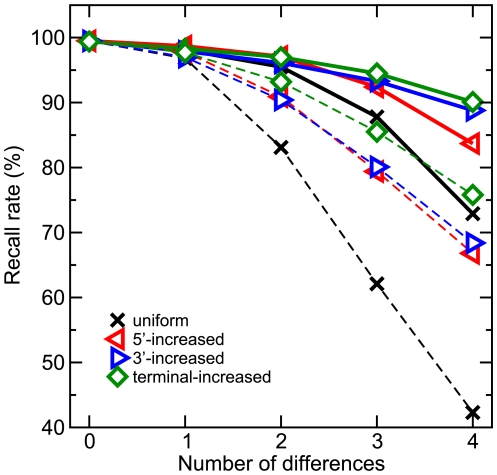
With few exceptions, current methods for short read mapping make use of simple seed heuristics to speed up the search. Most of the underlying matching models neglect the necessity to allow not only mismatches, but also insertions and deletions. Current evaluations indicate, however, that very different error models apply to the novel high-throughput sequencing methods. While the most frequent error-type in Illumina reads are mismatches, reads produced by 454's GS FLX predominantly contain insertions and deletions (indels). Even though 454 sequencers are able to produce longer reads, the method is frequently applied to small RNA (miRNA and siRNA) sequencing. Fast and accurate matching in particular of short reads with diverse errors is therefore a pressing practical problem. We introduce a matching model for short reads that can, besides mismatches, also cope with indels. It addresses different error models. For example, it can handle the problem of leading and trailing contaminations caused by primers and poly-A tails in transcriptomics or the length-dependent increase of error rates. In these contexts, it thus simplifies the tedious and error-prone trimming step. For efficient searches, our method utilizes index structures in the form of enhanced suffix arrays. In a comparison with current methods for short read mapping, the presented approach shows significantly increased performance not only for 454 reads, but also for Illumina reads. Our approach is implemented in the software segemehl available at http://www.bioinf.uni-leipzig.de/Software/segemehl/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
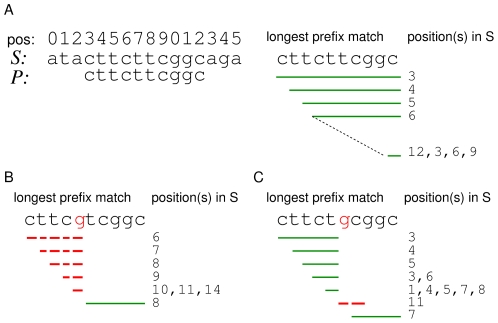
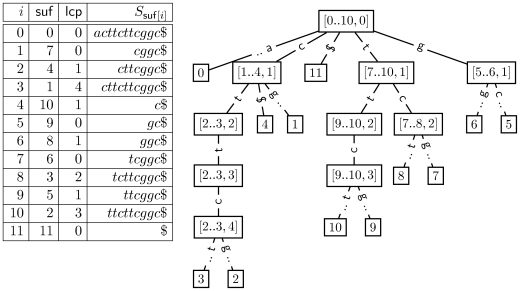
 denote the matching stem for Pi = ith suffix of P. The qth interval in
denote the matching stem for Pi = ith suffix of P. The qth interval in  , denoted by
, denoted by  , implicitly represents the set of suffixes in S matching P[i‥i+q−1]. The path for the first suffix P
0 is of length two (green solid line). Hence, the equivalent matching stem
, implicitly represents the set of suffixes in S matching P[i‥i+q−1]. The path for the first suffix P
0 is of length two (green solid line). Hence, the equivalent matching stem  is a sequence of three intervals:
is a sequence of three intervals:  ,
,  and
and  . Since
. Since  only represents the suffix S
7, the longest prefix match of P
0 is of length 2 occurring at position 7 of the reference sequence (right panel). The matching stem
only represents the suffix S
7, the longest prefix match of P
0 is of length 2 occurring at position 7 of the reference sequence (right panel). The matching stem  for P
1 (red solid line) ends with
for P
1 (red solid line) ends with  . Therefore, matches of length one occur at positions 8 and 9 in S. The longest prefix match for P
3 occurs at position 6 of S (dashed orange line). Note, that the intervals
. Therefore, matches of length one occur at positions 8 and 9 in S. The longest prefix match for P
3 occurs at position 6 of S (dashed orange line). Note, that the intervals  of
of  equivalently represent S
6. An alternative path leads to a match with position 4. The branch
equivalently represent S
6. An alternative path leads to a match with position 4. The branch  denotes the alternative that accepts the mismatch of g and t at position 1 of P
0.
denotes the alternative that accepts the mismatch of g and t at position 1 of P
0.
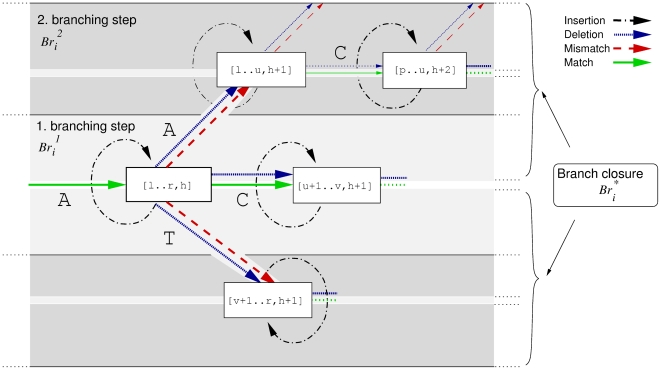
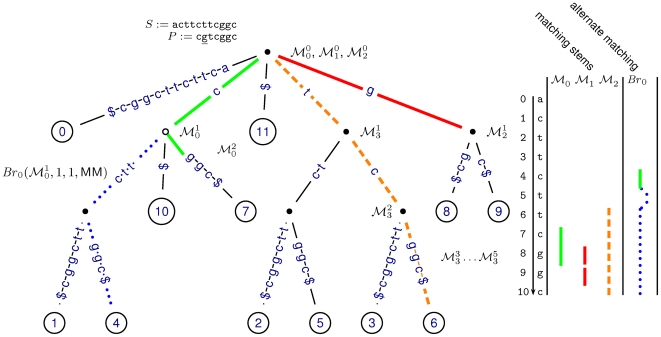
 of length h, is split into its children [l‥u, h+1], [u+1‥v, h+1] and [v+1‥r, h+1] by matching an additional character a∈{A, C, T}. We proceed building
of length h, is split into its children [l‥u, h+1], [u+1‥v, h+1] and [v+1‥r, h+1] by matching an additional character a∈{A, C, T}. We proceed building  by matching the character C (solid bold green line). Beforehand, alternative suffix intervals are stored in
by matching the character C (solid bold green line). Beforehand, alternative suffix intervals are stored in  , either representing mismatches (dashed red line), insertions (dashed dotted black line) or deletions (dotted blue line).
, either representing mismatches (dashed red line), insertions (dashed dotted black line) or deletions (dotted blue line).  holds suffix link intervals that in turn branch off from
holds suffix link intervals that in turn branch off from  . The branch closure
. The branch closure  holds all such alternative intervals.
holds all such alternative intervals.
References
-
- Myers G. A fast bit-vector algorithm for approximate string matching based on dynamic programming. J ACM. 1999;46:395–415.
-
- Abouelhoda MI, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. J Discr Algorithms. 2004;2:53–86.
-
- Rothberg JM, Leamon JH. The development and impact of 454 sequencing. Nat Biotechnol. 2008;26:1117–1124. - PubMed
-
- Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
