

Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane
- PMID: 19753315
- PMCID: PMC2742642
Disease-causing mutations in the CLRN1 gene alter normal CLRN1 protein trafficking to the plasma membrane
Abstract
Purpose: Mutations of clarin 1 (CLRN1) cause Usher syndrome type 3 (USH3). To determine the effects of USH3 mutations on CLRN1 function, we examined the cellular distribution and stability of both normal and mutant CLRN1 in vitro. We also searched for novel disease-causing mutations in a cohort of 59 unrelated Canadian and Finnish USH patients.
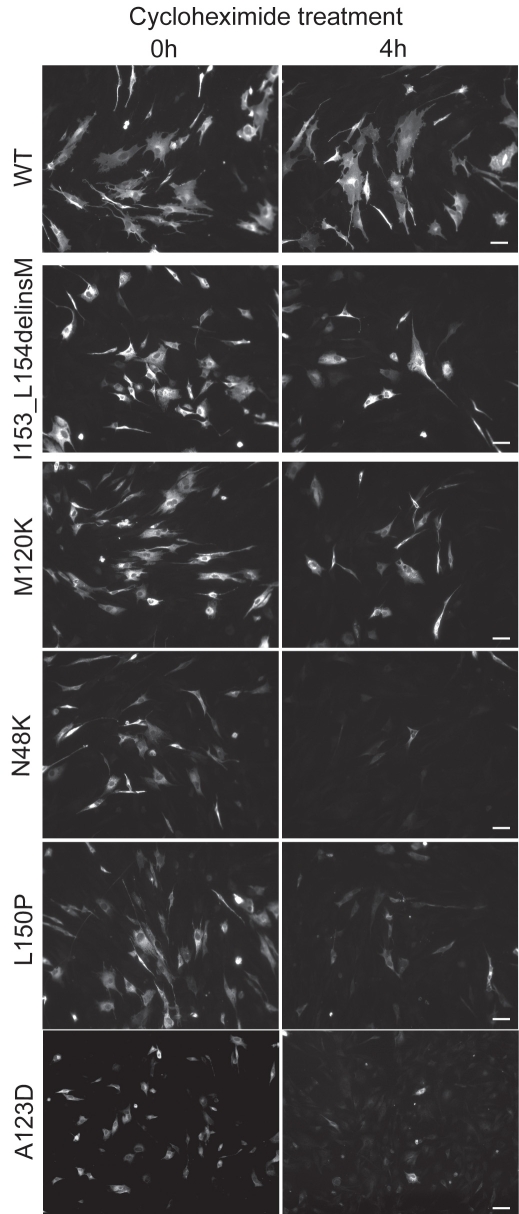
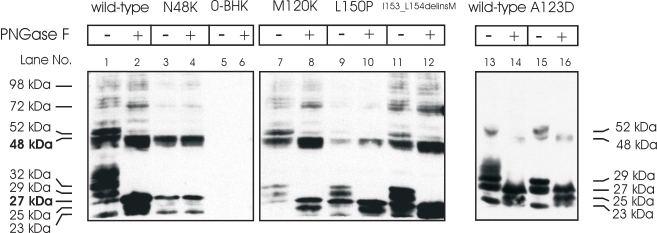
Methods: Mutation screening was performed by DNA sequencing. For the functional studies, wild-type (WT) and mutant CLRN1 genes were expressed as hemagglutinin (HA) tagged fusion proteins by transient transfection of BHK-21 cells. Subcellular localization of CLRN1-HA was examined by confocal microscopy. The N-glycosylation status of CLRN1 was studied by using the N-glycosidase F (PNGase F) enzyme and western blotting. Cycloheximide treatment was used to assess the stability of CLRN1 protein.
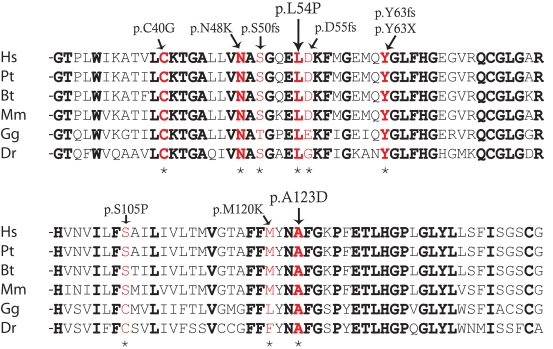
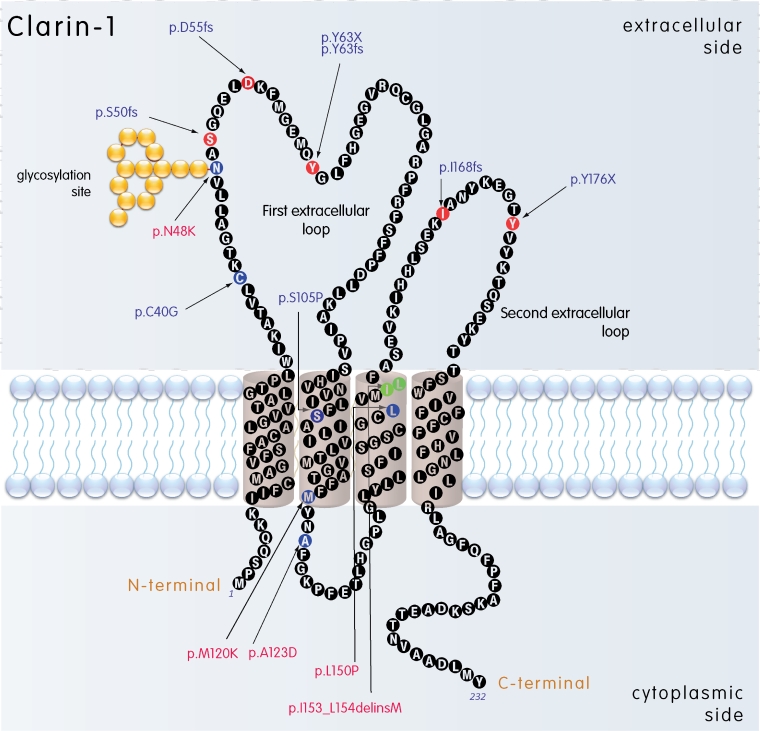
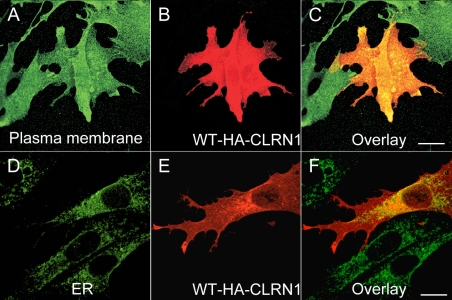
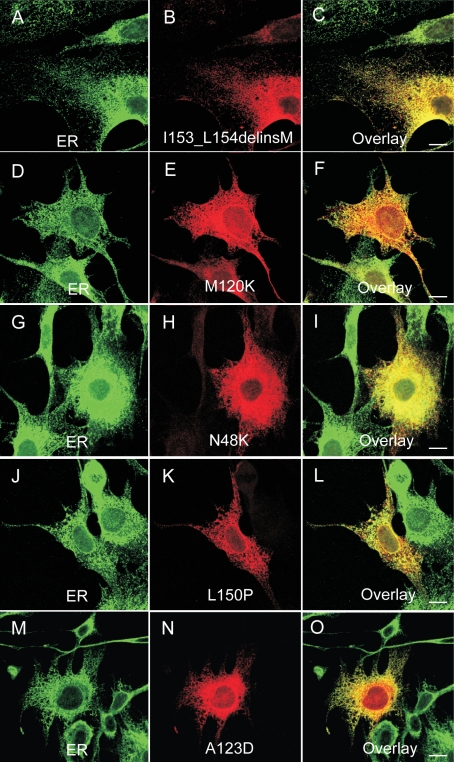
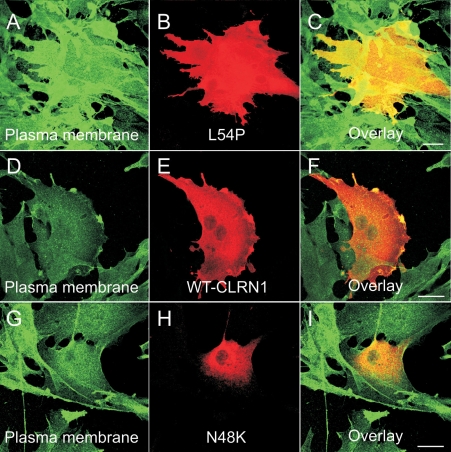
Results: We found three previously reported pathogenic mutations, p.A123D, p.N48K, and p.Y176X, and a novel sequence variant, p.L54P, from the studied USH patients. The WT HA-tagged CLRN1 was correctly trafficked to the plasma membrane, whereas mutant CLRN1-HA proteins were mislocalized and retained in the endoplasmic reticulum. PNGase F treatment of CLRN1-HA resulted in an electrophoretic mobility shift consistent with sugar residue cleavage in WT and in all CLRN1 mutants except in p.N48K mutated CLRN1, in which the mutation abolishes the glycosylation site. Inhibition of protein expression with cycloheximide indicated that WT CLRN1-HA remained stable. In contrast, the CLRN1 mutants showed reduced stability.
Conclusions: WT CLRN1 is a glycoprotein localized to the plasma membrane in transfected BHK-21 cells. Mutant CLRN1 proteins are mislocalized. We suggest that part of the pathogenesis of USH3 may be associated with defective intracellular trafficking as well as decreased stability of mutant CLRN1 proteins.
Figures
References
-
- Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet. 2001;2:271–97. - PubMed
-
- Ahmed ZM, Riazuddin S, Wilcox ER. The molecular genetics of Usher syndrome. Clin Genet. 2003;63:431–44. - PubMed
-
- Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83:97–119. - PubMed
-
- Kremer H, van Wijk E, Märker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15:R262–70. - PubMed
-
- Spandau UH, Rohrschneider K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch Clin Exp Ophthalmol. 2002;240:495–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical