

Extraction of human kinase mutations from literature, databases and genotyping studies
- PMID: 19758464
- PMCID: PMC2745582
- DOI: 10.1186/1471-2105-10-S8-S1
Extraction of human kinase mutations from literature, databases and genotyping studies
Abstract
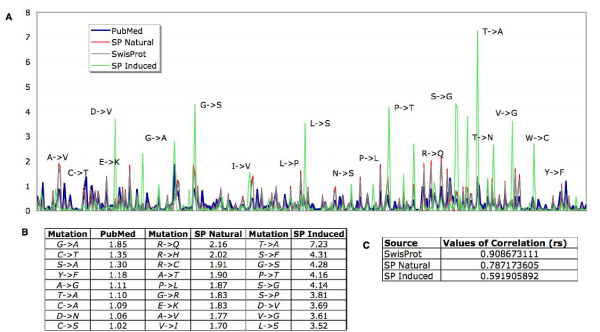
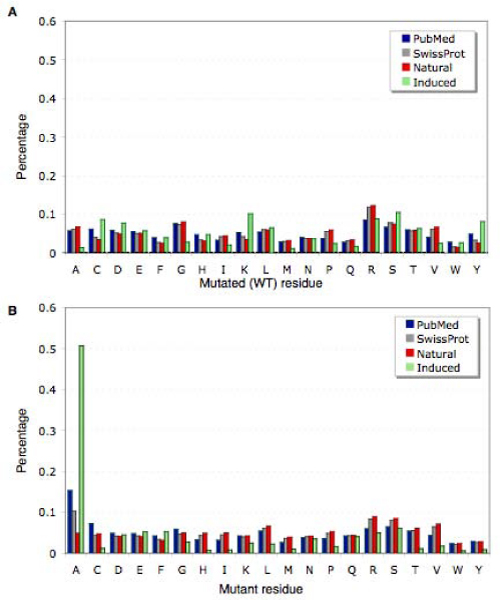
Background: There is a considerable interest in characterizing the biological role of specific protein residue substitutions through mutagenesis experiments. Additionally, recent efforts related to the detection of disease-associated SNPs motivated both the manual annotation, as well as the automatic extraction, of naturally occurring sequence variations from the literature, especially for protein families that play a significant role in signaling processes such as kinases. Systematic integration and comparison of kinase mutation information from multiple sources, covering literature, manual annotation databases and large-scale experiments can result in a more comprehensive view of functional, structural and disease associated aspects of protein sequence variants. Previously published mutation extraction approaches did not sufficiently distinguish between two fundamentally different variation origin categories, namely natural occurring and induced mutations generated through in vitro experiments.
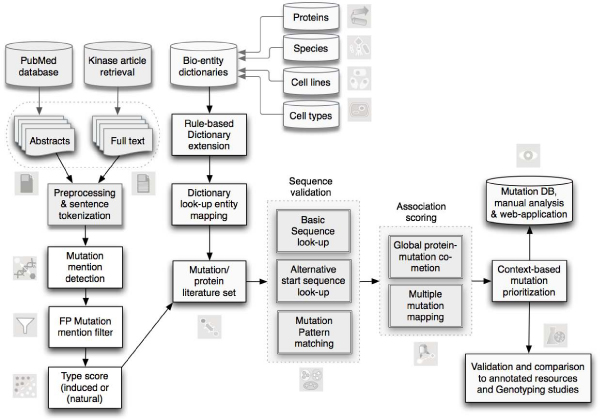
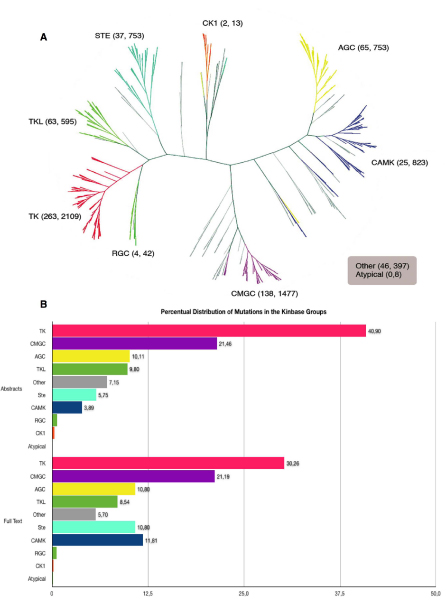
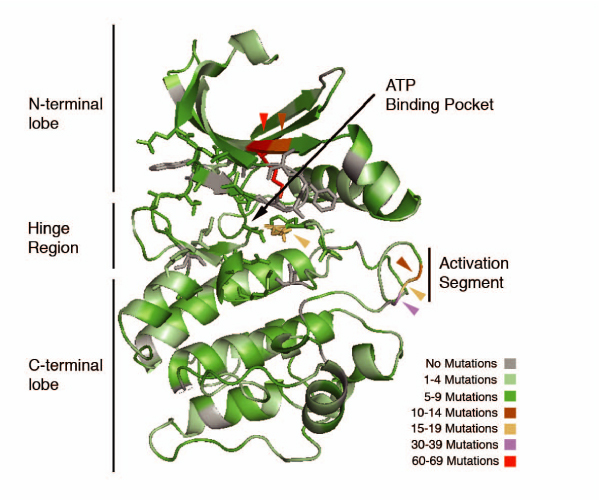
Results: We present a literature mining pipeline for the automatic extraction and disambiguation of single-point mutation mentions from both abstracts as well as full text articles, followed by a sequence validation check to link mutations to their corresponding kinase protein sequences. Each mutation is scored according to whether it corresponds to an induced mutation or a natural sequence variant. We were able to provide direct literature links for a considerable fraction of previously annotated kinase mutations, enabling thus more efficient interpretation of their biological characterization and experimental context. In order to test the capabilities of the presented pipeline, the mutations in the protein kinase domain of the kinase family were analyzed. Using our literature extraction system, we were able to recover a total of 643 mutations-protein associations from PubMed abstracts and 6,970 from a large collection of full text articles. When compared to state-of-the-art annotation databases and high throughput genotyping studies, the mutation mentions extracted from the literature overlap to a good extent with the existing knowledgebases, whereas the remaining mentions suggest new mutation records that were not previously annotated in the databases.
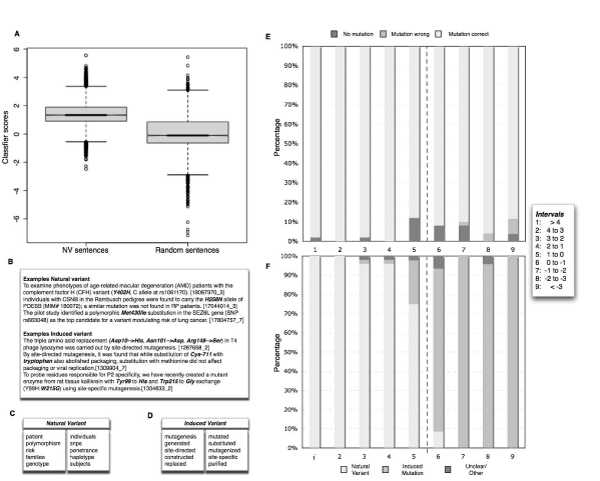
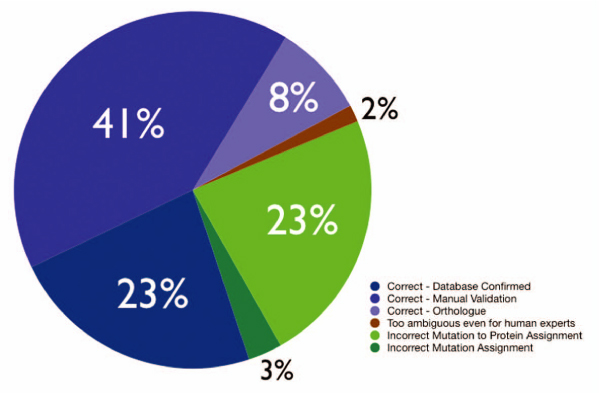
Conclusion: Using the proposed residue disambiguation and classification approach, we were able to differentiate between natural variant and mutagenesis types of mutations with an accuracy of 93.88. The resulting system is useful for constructing a Gold Standard set of mutations extracted from the literature by human experts with minimal manual curation effort, providing direct pointers to relevant evidence sentences. Our system is able to recover mutations from the literature that are not present in state-of-the-art databases. Human expert manual validation of a subset of the literature extracted mutations conducted on 100 mutations from PubMed abstracts highlights that almost three quarters (72%) of the extracted mutations turned out to be correct, and more than half of these had not been previously annotated in databases.
Figures
References
-
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298:1912–1934. - PubMed
-
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the Cyclin-dependent Kinase Cdk1. Nature. 2003;425:859–864. - PubMed
-
- Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJR, Stern DF, De Virgilio C, Tyers M, Andrews B, Gerstein M, Schweitzer B, Predki PF, Snyder M. Global Analysis of Protein Phosphorylation in Yeast. Nature. 2005;438:679–684. - PubMed
-
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. - PubMed
-
- Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263–74. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
