

Control of herpes simplex virus replication is mediated through an interferon regulatory factor 3-dependent pathway
- PMID: 19759149
- PMCID: PMC2786705
- DOI: 10.1128/JVI.00888-09
Control of herpes simplex virus replication is mediated through an interferon regulatory factor 3-dependent pathway
Abstract
The type I interferon (IFN) cascade is critical in controlling viral replication and pathogenesis. Recognition pathways triggered by viral infection rapidly induce the type I IFN cascade, often in an IFN regulatory factor 3 (IRF-3)-dependent fashion. This dependence predicts that loss of IRF-3 would render early recognition pathways inoperative and thereby impact virus replication, but this has not been observed previously with herpes simplex virus type 1 (HSV-1) in vitro. In this study, HSV-1-infected IRF-3(-/-) bone marrow-derived dendritic cells (BMDCs) and macrophages supported increased HSV replication compared to control cells. In addition, IRF-3-deficient BMDCs exhibited delayed type I IFN synthesis compared to control cells. However, while IFN pretreatment of IRF-3(-/-) BMDCs resulted in reduced virus titers, a far greater reduction was seen after IFN treatment of wild-type cells. This suggests that even in the presence of exogenously supplied IFN, IRF-3(-/-) BMDCs are inherently defective in the control of HSV-1 replication. Together, these results demonstrate a critical role for IRF-3-mediated pathways in controlling HSV-1 replication in cells of the murine immune system.
Figures

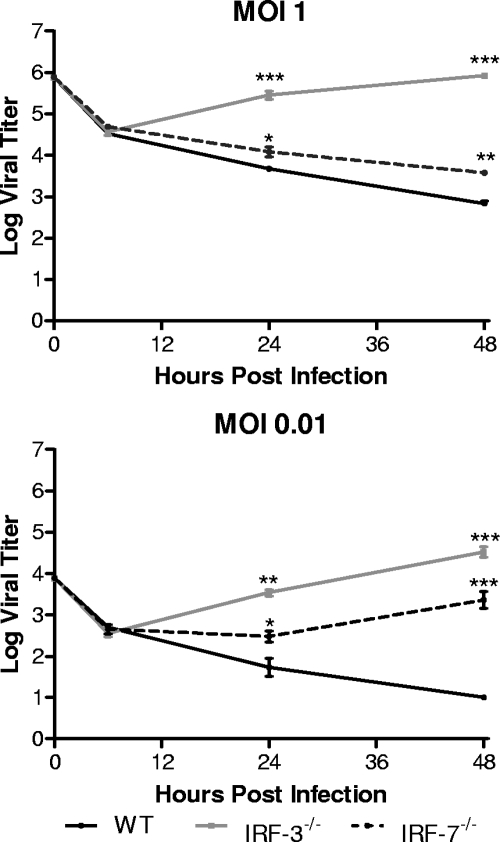



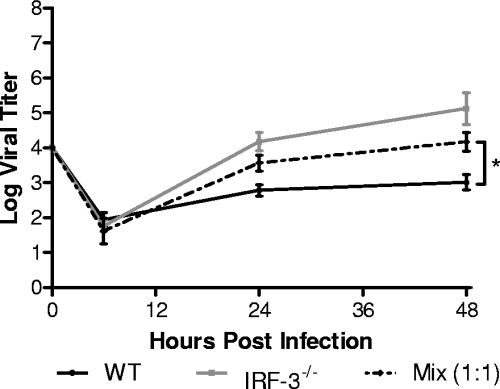


References
-
- Andersen, J., S. VanScoy, T. F. Cheng, D. Gomez, and N. C. Reich. 2008. IRF-3-dependent and augmented target genes during viral infection. Genes Immun. 9:168-175. - PubMed
-
- Bauer, D., S. Mrzyk, N. van Rooijen, K. P. Steuhl, and A. Heiligenhaus. 2000. Macrophage-depletion influences the course of murine HSV-1 keratitis. Curr. Eye Res. 20:45-53. - PubMed
-
- Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919-923. - PubMed
-
- Cheng, H., T. M. Tumpey, H. F. Staats, N. van Rooijen, J. E. Oakes, and R. N. Lausch. 2000. Role of macrophages in restricting herpes simplex virus type 1 growth after ocular infection. Investig. Ophthalmol. Vis. Sci. 41:1402-1409. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
