

doi: 10.1186/gb-2009-10-9-r98.
Epub 2009 Sep 17.
Simultaneous alignment of short reads against multiple genomes
Affiliations
- PMID: 19761611
- PMCID: PMC2768987
- DOI: 10.1186/gb-2009-10-9-r98
Item in Clipboard
Simultaneous alignment of short reads against multiple genomes
Genome Biol.
2009.
Abstract
Genome resequencing with short reads generally relies on alignments against a single reference. GenomeMapper supports simultaneous mapping of short reads against multiple genomes by integrating related genomes (e.g., individuals of the same species) into a single graph structure. It constitutes the first approach for handling multiple references and introduces representations for alignments against complex structures. Demonstrated benefits include access to polymorphisms that cannot be identified by alignments against the reference alone. Download GenomeMapper at http://1001genomes.org.
Figures
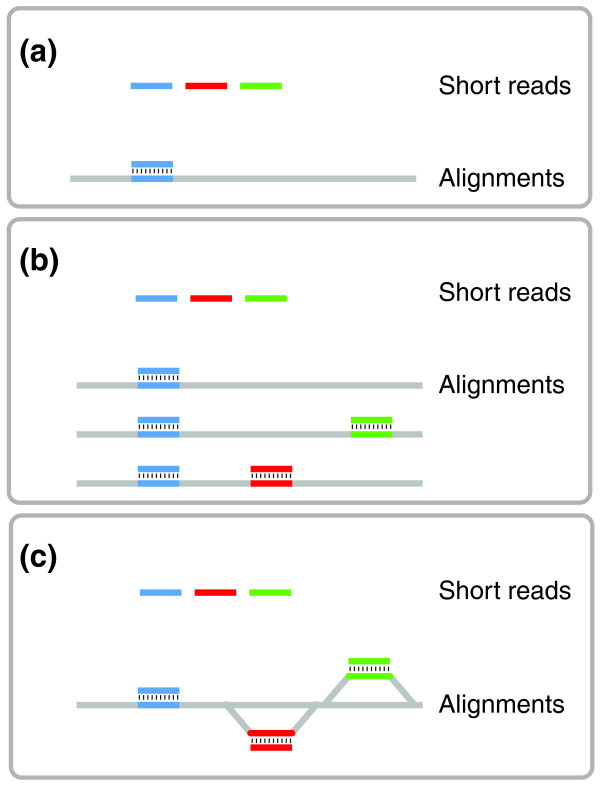
Efficient alignments against multiple genomes. (a) Only reads that are sufficiently similar can be aligned against a single reference. (b) Separate alignment against multiple genomes allows access to divergent regions, but results in redundant alignments of reads that match all targets (blue). (c) Alignments against a graph index representing multiple genomes provide access to divergent regions without redundant alignments.
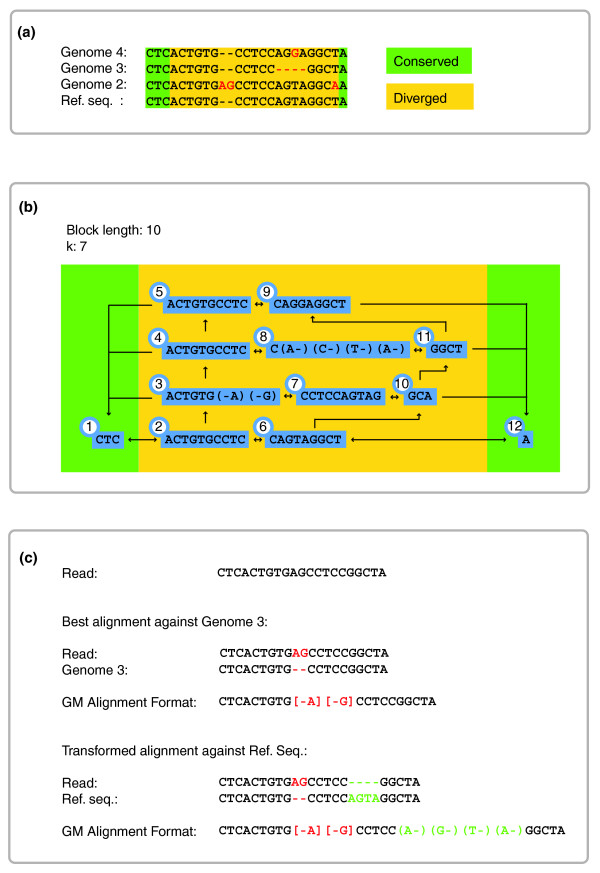
GenomeMapper's graph index structure. (a) Examples of orthologous sequences in four divergent genomes. Sequences at the beginning and end of each fragment are shared (underlaid with green boxes). Divergent regions start k-1 positions (in this case, six positions) before the first true variable position, to account for the k-mer length used for the hash-key calculation. (b) Graph structure created by these sequences, with k-mer length 7, and maximal block length of 10 (instead of 256) for reasons of illustration. The number attached to each block is its unique identifier. Note that blocks do not occupy their maximal block length after an indel, exemplified by blocks 3 and 8. Blocks 1 and 12 correspond to sequences identical in all four genomes and are present only once in the index structure. Arrows between the blocks visualize the edges between the nodes in the genome graph as they are stored in the block table [see Table S1 in Additional data file 1]. (c) Alignment of a read against the most similar genome, Genome 3, with a 2-bp insertion. Although the insertion also is observed in Genome 2, the 4-bp deletion downstream in Genome 3 makes the read more similar to it than to Genome 2. The transformed alignment of the read against the original reference sequence (Ref. seq.) includes the 4-bp deletion (as supported by Genome 3) given in parentheses (green), whereas the 2-bp insertion (which is supported neither by Genome 3 nor by the reference sequence) is annotated like a mismatch by using square brackets.
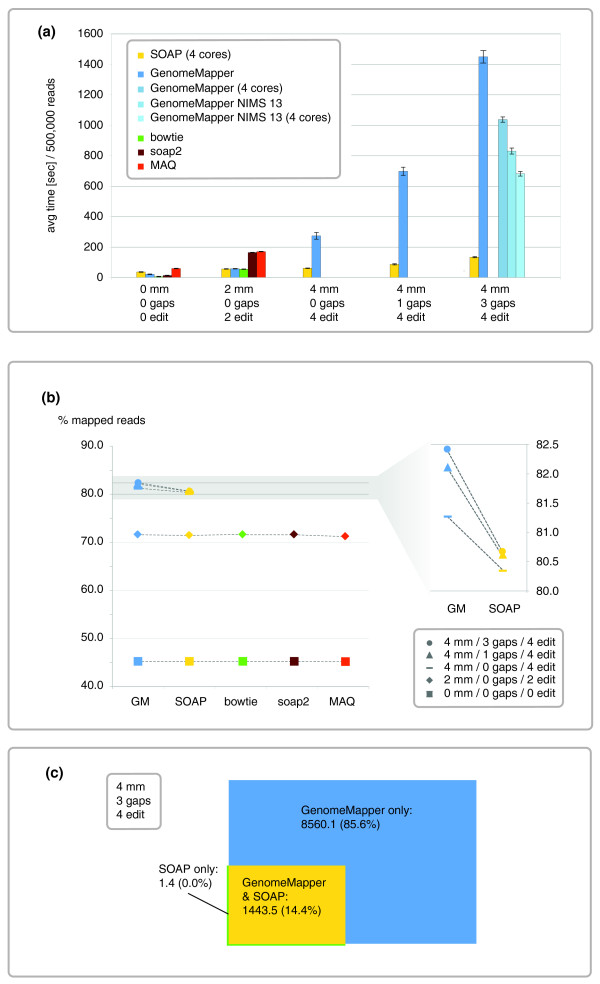
Performance of GenomeMapper compared with that of other short-read alignment tools. (a) Runtime, measured as wall clock time between invocation and termination of the program, averaged from 10 independent tests with different random sets of 500,000 short reads from Est-1. The worst test was excluded from average calculations. Error bars indicate standard deviation. mm, gaps, and edit refer to the maximal number of mismatches, gaps and edit operations allowed. GenomeMapper was run with four different parameter settings: the serial version; the parallel version on four cores; the serial version merely aligning NIMS of length 13 or longer; and the parallel version aligning only NIMS of length 13 or longer. SOAP was found running on up to four CPUs instead of only one CPU, as configured with the command line (option -p). (b) Average sensitivity, measured as the percentage of aligned reads. Only GenomeMapper and SOAP can perform gapped alignments. (c) Average sensitivity of alignments, allowing three gaps and four mismatches with a combined maximum of four edit operations, measured as number of reads with gapped alignments. Fractions refer to the number of all reads with gapped alignments.
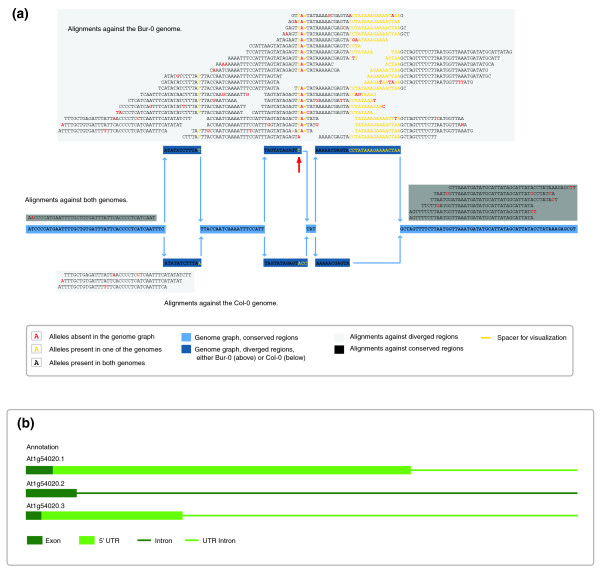
Alignments against a 17-bp insertion present in a nonreference genome. (a) Alignments of Est-1 reads against the graph of Arabidopsis chromosome 1, reference positions 20,166,584 to 20,166,747. Alignments against both the Col-0 reference and the Bur-0 variant genomes are highlighted in dark gray; alignments of reads aligning best against a single genome are highlighted in light gray. Most reads align against the Bur-0 allele, suggesting that Est-1 is more similar to Bur-0 at this locus. In particular, the 17-bp insertion found in Bur-0 is supported by the Est-1 reads. Because of the alignment constraints (maximum of four edit operations), these alignments could not have been performed against the Col-0 sequence only. Within the second divergent region, indicated by a red arrow, Bur-0 has a complex change, ACC->T, relative to Col-0, with Est-1 featuring a third allele, ACC->TA. Because this change is near the 17-bp insertion, only a subset of the alignments would have been found with single reference alignments only. For simplicity, Tsu-1, which also is included in the graph target, is not shown here. (b) Annotation of this region with respect to the Col-0 reference genome.
References
-
- Hillier LW, Marth GT, Quinlan AR, Dooling D, Fewell G, Barnett D, Fox P, Glasscock JI, Hickenbotham M, Huang W, Magrini VJ, Richt RJ, Sander SN, Stewart DA, Stromberg M, Tsung EF, Wylie T, Schedl T, Wilson RK, Mardis ER. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. - DOI - PubMed
-
- Ahn SM, Kim TH, Lee S, Kim D, Ghang H, Kim DS, Kim BC, Kim SY, Kim WY, Kim C, Park D, Lee YS, Kim S, Reja R, Jho S, Kim CG, Cha JY, Kim KH, Lee B, Bhak J, Kim SJ. The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome Res. 2009;19:1622–1629. doi: 10.1101/gr.092197.109. - DOI - PMC - PubMed
-
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, Boutell JM, Bryant J, Carter RJ, Keira Cheetham R, Cox AJ, Ellis DJ, Flatbush MR, Gormley NA, Humphray SJ, Irving LJ, Karbelashvili MS, Kirk SM, Li H, Liu X, Maisinger KS, Murray LJ, Obradovic B, Ost T, Parkinson ML, Pratt MR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. - DOI - PMC - PubMed
-
- Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, Mudge J, Miller NA, Hong D, Bell CJ, Kim HS, Chung IS, Lee WC, Lee JS, Seo SH, Yun JY, Woo HN, Lee H, Suh D, Kim HJ, Yavartanoo M, Kwak M, Zheng Y, Lee MK, Kim JY, Gokcumen O, Mills RE, Zaranek AW, Thakuria J, Wu X, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
