

Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress
- PMID: 19761620
- PMCID: PMC2761850
- DOI: 10.1186/1743-8977-6-24
Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress
Abstract
Air Pollution has been associated with significant adverse health effects leading to increased morbidity and mortality. Cumulative epidemiological and experimental data have shown that exposure to air pollutants lead to increased cardiovascular ischemic events and enhanced atherosclerosis. It appears that these associations are much stronger with the air particulate matter (PM) component and that in urban areas, the smaller particles could be more pathogenic, as a result of their greater propensity to induce systemic prooxidant and proinflammatory effects. Much is still unknown about the toxicology of ambient particulates as well as the pathogenic mechanisms responsible for the induction of adverse cardiovascular health effects. It is expected that better understanding of these effects will have large implications and may lead to the formulation and implementation of new regulatory policies. Indeed, we have found that ultrafine particles (<0.18 mum) enhance early atherosclerosis, partly due to their high content in redox cycling chemicals and their ability to synergize with known proatherogenic mediators in the promotion of tissue oxidative stress. These changes take place in parallel with increased evidence of phase 2 enzymes expression, via the electrophile-sensitive transcription factor, p45-NFE2 related transcription factor 2 (Nrf2). Exposure to ultrafine particles also results in alterations of the plasma HDL anti-inflammatory function that could be indicative of systemic proatherogenic effects. This article reviews the epidemiological, clinical and experimental animal evidence that support the association of particulate matter with atherogenesis. It also discusses the possible pathogenic mechanisms involved, the physicochemical variables that may be of importance in the greater toxicity exhibited by a small particle size, interaction with genes and other proatherogenic factors as well as important elements to consider in the design of future mechanistic studies.Extensive epidemiological evidence supports the association of air pollution with adverse health effects 123. It is increasingly being recognized that such effects lead to enhanced morbidity and mortality, mostly due to exacerbation of cardiovascular diseases and predominantly those of ischemic character 4. Indeed, in addition to the classical risk factors such as serum lipids, smoking, hypertension, aging, gender, family history, physical inactivity and diet, recent data have implicated air pollution as an important additional risk factor for atherosclerosis. This has been the subject of extensive reviews 56 and a consensus statement from the American Heart Association 7. This article reviews the supporting epidemiological and animal data, possible pathogenic mechanisms and future perspectives.
Figures
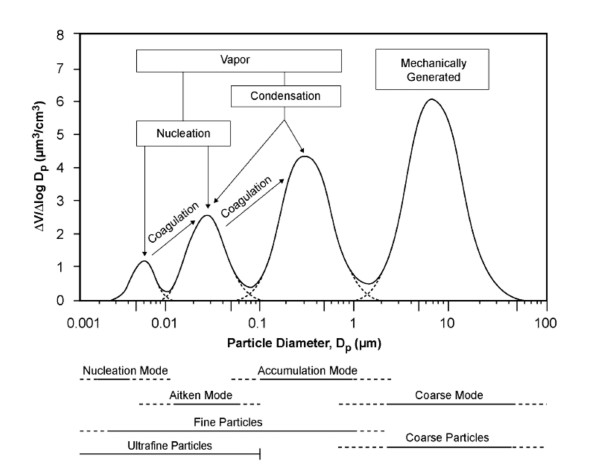
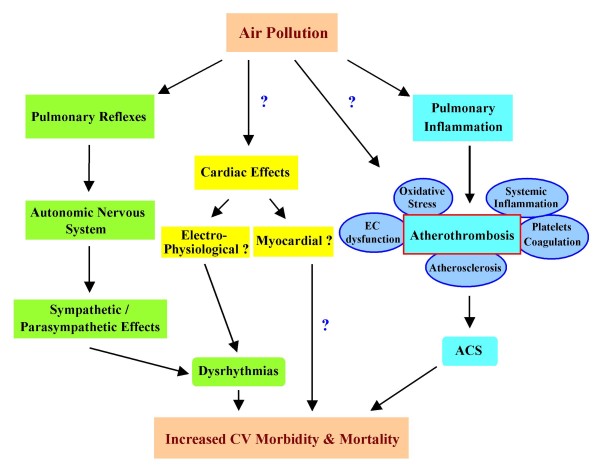
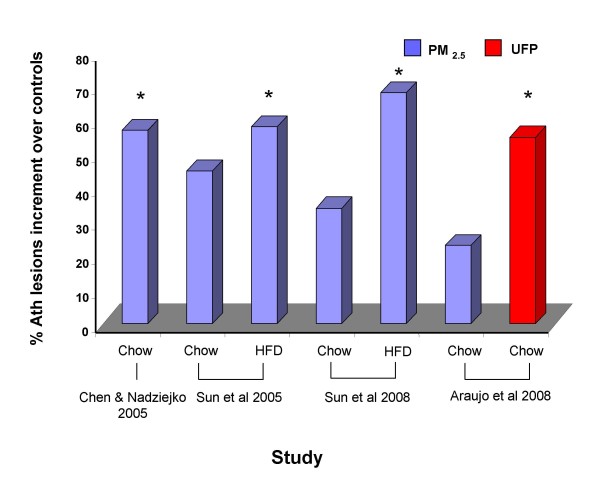
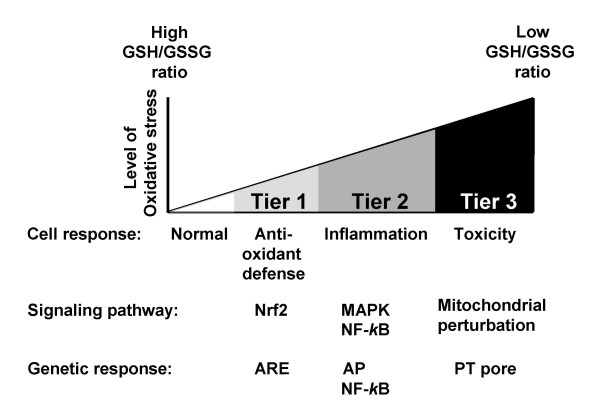
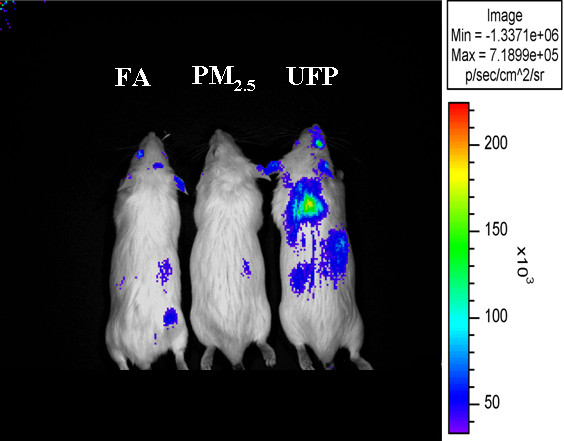
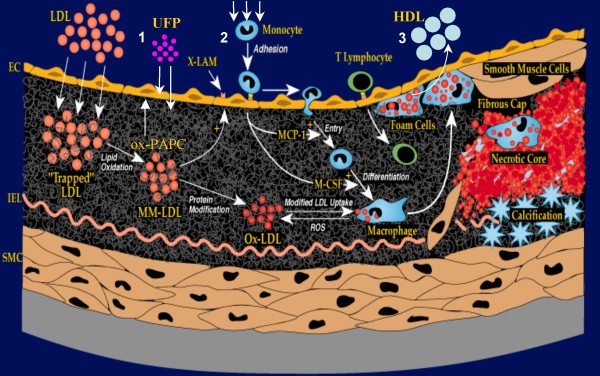
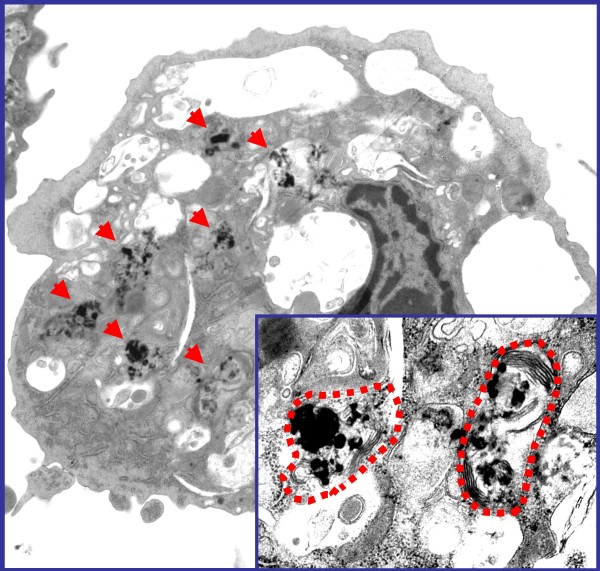
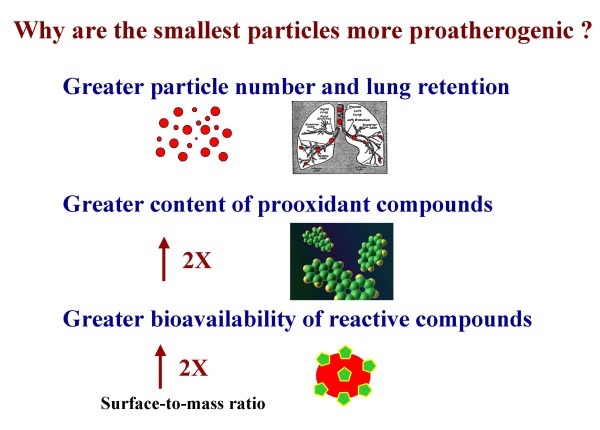
References
-
- Pope CA, 3rd, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, Heath CW., Jr Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am J Respir Crit Care Med. 1995;151:669–674. - PubMed
-
- Pope CA, 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. - DOI - PubMed
-
- Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Circ Res. 2006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
