

A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects
- PMID: 19762328
- PMCID: PMC2981023
- DOI: 10.1136/jmg.2009.069997
A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects
Abstract
Background: Ostium secundum atrial septal defects (ASDII) account for approximately 10% of all congenital heart defects (CHD), and mutations in cardiac transcription factors, including TBX20, were identified as an underlying cause for ASDII. However, very little is known about disease penetrance in families and functional consequences of inherited TBX20 mutations.
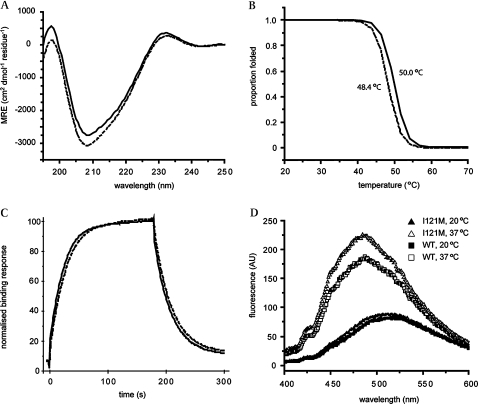
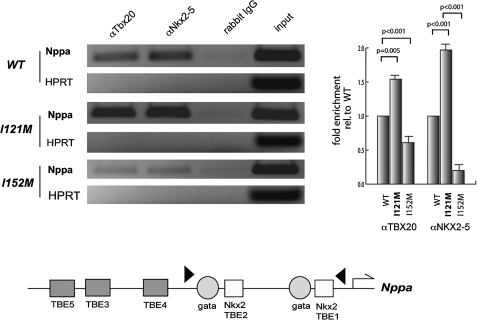
Methods: The coding region of TBX20 was directly sequenced in 170 ASDII patients. Functional consequences of one novel mutation were investigated by surface plasmon resonance, CD spectropolarymetry, fluorescence spectrophotometry, luciferase assay and chromatin immunoprecipitation.
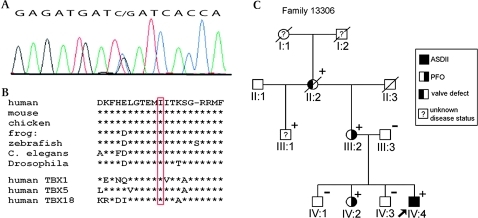
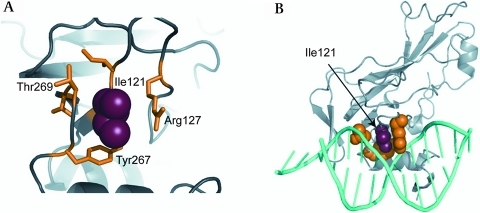
Results: We found a novel mutation in a highly conserved residue in the T-box DNA binding domain (I121M) segregating with CHD in a three generation kindred. Four mutation carriers revealed cardiac phenotypes in terms of cribriform ASDII, large patent foramen ovale or cardiac valve defects. Interestingly, tertiary hydrophobic interactions within the mutant TBX20 T-box were significantly altered leading to a more dynamic structure of the protein. Moreover, Tbx20-I121M resulted in a significantly enhanced transcriptional activity, which was further increased in the presence of co-transcription factors GATA4/5 and NKX2-5. Occupancy of DNA binding sites on target genes was also increased.
Conclusions: We suggest that TBX20-I121M adopts a more fluid tertiary structure leading to enhanced interactions with cofactors and more stable transcriptional complexes on target DNA sequences. Our data, combined with that of others, suggest that human ASDII may be related to loss-of-function as well as gain-of-function TBX20 mutations.
Conflict of interest statement
Figures

References
-
- Bruneau BG. The developmental genetics of congenital heart disease. Nature 2008;451:943–8 - PubMed
-
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003;424:443–7 - PubMed
-
- Kasahara H, Benson DW. Biochemical analyses of eight NKX2.5 homeodomain missense mutations causing atrioventricular block and cardiac anomalies. Cardiovasc Res 2004;64:40–51 - PubMed
-
- Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat 2006;27:293–4 - PubMed
-
- Caputo S, Capozzi G, Russo MG, Esposito T, Martina L, Cardaropoli D, Ricci C, Argiento P, Pacileo G, Calabro R. Familial recurrence of congenital heart disease in patients with ostium secundum atrial septal defect. Eur Heart J 2005;26:2179–84 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases