

Mutated K-ras(Asp12) promotes tumourigenesis in Apc(Min) mice more in the large than the small intestines, with synergistic effects between K-ras and Wnt pathways
- PMID: 19765110
- PMCID: PMC2768154
- DOI: 10.1111/j.1365-2613.2009.00667.x
Mutated K-ras(Asp12) promotes tumourigenesis in Apc(Min) mice more in the large than the small intestines, with synergistic effects between K-ras and Wnt pathways
Abstract
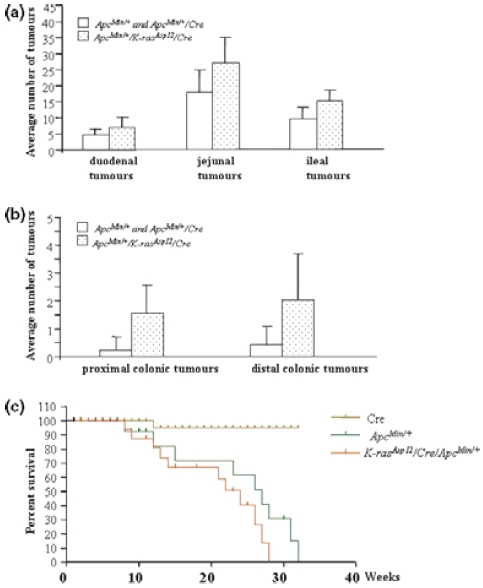
Summary K-ras mutations are found in 40-50% of human colorectal adenomas and carcinomas, but their functional contribution remains incompletely understood. Here, we show that a conditional mutant K-ras mouse model (K-ras(Asp12)/Cre), with transient intestinal Cre activation by beta-Naphthoflavone (beta-NF) treatment, displayed transgene recombination and K-ras(Asp12) expression in the murine intestines, but developed few intestinal adenomas over 2 years. However, when crossed with Apc(Min/+) mice, the K-ras(Asp12)/Cre/Apc(Min/+) offspring showed acceleration of intestinal tumourigenesis with significantly changed average lifespan (P < 0.05) decreased to 18.4 +/- 5.4 weeks from 20.9 +/- 4.7 weeks (control Apc(Min/+) mice). The numbers of adenomas in the small intestine and large intestine were significantly (P < 0.01) increased by 1.5-fold and 5.7-fold, respectively, in K-ras(Asp12)/Cre/Apc(Min/+) mice compared with Apc(Min/+) mice, with the more marked increase in adenoma prevalence in the large intestine. To explore possible mechanisms for K-ras(Asp12) and Apc(Min) co-operation, the Mitogen-activated protein kinase (Mapk), Akt and Wnt signalling pathways, including selected target gene expression levels, were evaluated in normal large intestine and large intestinal tumours. K-ras(Asp12) increased activation of Mapk and Akt signalling pathway targets phospho-extracellular signal-regulated kinase (pErk) and pAkt, and increased relative expression levels of Wnt pathway targets vascular endothelial growth factor (VEGF), gastrin, cyclo-oxygenase 2 (Cox2) and T-cell lymphoma invasion and metastasis 1 (Tiam1) in K-ras(Asp12)/Cre/Apc(Min/+) adenomas compared with that of Apc(Min/+) adenomas, although other Wnt signalling pathway target genes such as Peroxisome proliferator-activated receptor delta (PPARd), matrix metalloproteinase 7 (MMP7), protein phosphatase 1 alpha (PP1A) and c-myc remained unchanged. In conclusion, intestinal expression of K-ras(Asp12) promotes mutant Apc-initiated intestinal adenoma formation in vivo more in the large intestine than the small intestine, with evidence of synergistic co-operation between mutant K-ras and Apc involving increased expression of some Wnt-pathway target genes.
Figures





Similar articles
-
Synergism between K-rasVal12 and mutant Apc accelerates murine large intestinal tumourigenesis.Oncol Rep. 2011 Jul;26(1):125-33. doi: 10.3892/or.2011.1288. Epub 2011 Apr 29. Oncol Rep. 2011. PMID: 21573497
-
Conditional expression of mutated K-ras accelerates intestinal tumorigenesis in Msh2-deficient mice.Oncogene. 2007 Jun 28;26(30):4415-27. doi: 10.1038/sj.onc.1210231. Epub 2007 Feb 5. Oncogene. 2007. PMID: 17297472
-
Mutant K-ras promotes carcinogen-induced murine colorectal tumourigenesis, but does not alter tumour chromosome stability.J Pathol. 2011 Feb;223(3):390-9. doi: 10.1002/path.2790. Epub 2010 Nov 11. J Pathol. 2011. PMID: 21171084
-
Interaction of the Wnt/β-catenin and RAS-ERK pathways involving co-stabilization of both β-catenin and RAS plays important roles in the colorectal tumorigenesis.Adv Biol Regul. 2018 May;68:46-54. doi: 10.1016/j.jbior.2018.01.001. Epub 2018 Jan 10. Adv Biol Regul. 2018. PMID: 29449169 Review.
-
Murine models of colorectal cancer: studying the role of oncogenic K-ras.Cell Mol Life Sci. 2003 Mar;60(3):495-506. doi: 10.1007/s000180300041. Cell Mol Life Sci. 2003. PMID: 12737309 Free PMC article. Review.
Cited by
-
Genomic instability and colon carcinogenesis: from the perspective of genes.Front Oncol. 2013 May 21;3:130. doi: 10.3389/fonc.2013.00130. eCollection 2013. Front Oncol. 2013. PMID: 23734346 Free PMC article.
-
Animal models of gastrointestinal and liver diseases. New mouse models for studying dietary prevention of colorectal cancer.Am J Physiol Gastrointest Liver Physiol. 2014 Aug 1;307(3):G249-59. doi: 10.1152/ajpgi.00019.2014. Epub 2014 May 29. Am J Physiol Gastrointest Liver Physiol. 2014. PMID: 24875098 Free PMC article. Review.
-
Activation of K-RAS by co-mutation of codons 19 and 20 is transforming.J Mol Signal. 2011 Mar 3;6:2. doi: 10.1186/1750-2187-6-2. J Mol Signal. 2011. PMID: 21371307 Free PMC article.
-
Inducible Intestine-Specific Expression of krasV12 Triggers Intestinal Tumorigenesis In Transgenic Zebrafish.Neoplasia. 2018 Dec;20(12):1187-1197. doi: 10.1016/j.neo.2018.10.002. Epub 2018 Nov 1. Neoplasia. 2018. PMID: 30390498 Free PMC article.
-
Human Colorectal Cancer from the Perspective of Mouse Models.Genes (Basel). 2019 Oct 11;10(10):788. doi: 10.3390/genes10100788. Genes (Basel). 2019. PMID: 31614493 Free PMC article. Review.
References
-
- Al-Aynati MM, Radulovich N, Riddell RH, Tsao MS. Epithelial-cadherin and beta-catenin expression changes in pancreatic intraepithelial neoplasia. Clin. Cancer Res. 2004;10:1235–1240. - PubMed
-
- Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J. Natl. Cancer Inst. 1998;90:675–684. - PubMed
-
- Araki Y, Okamura S, Hussain SP, et al. Regulation of cyclooxygenase-2 expression by the Wnt and ras pathways. Cancer Res. 2003;63:728–734. - PubMed
-
- Arends MJ, Frayling I. Mismatch repair deficiency in hereditary and sporadic colorectal cancer. In: Cunningham D, Topham C, Miles A, editors. The Effective Management of Colorectal Cancer. 4th edn. London: Aesculapius Medical Press; 2005. pp. 25–40. pp Chapter 2.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
