

I-TASSER: fully automated protein structure prediction in CASP8
- PMID: 19768687
- PMCID: PMC2782770
- DOI: 10.1002/prot.22588
I-TASSER: fully automated protein structure prediction in CASP8
Abstract
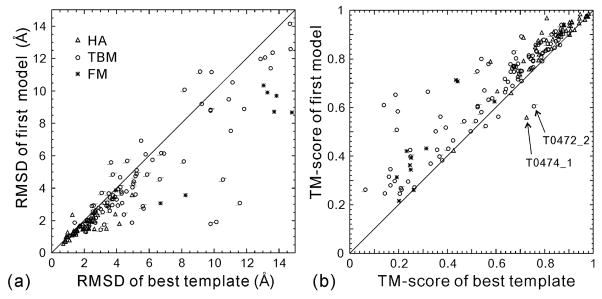
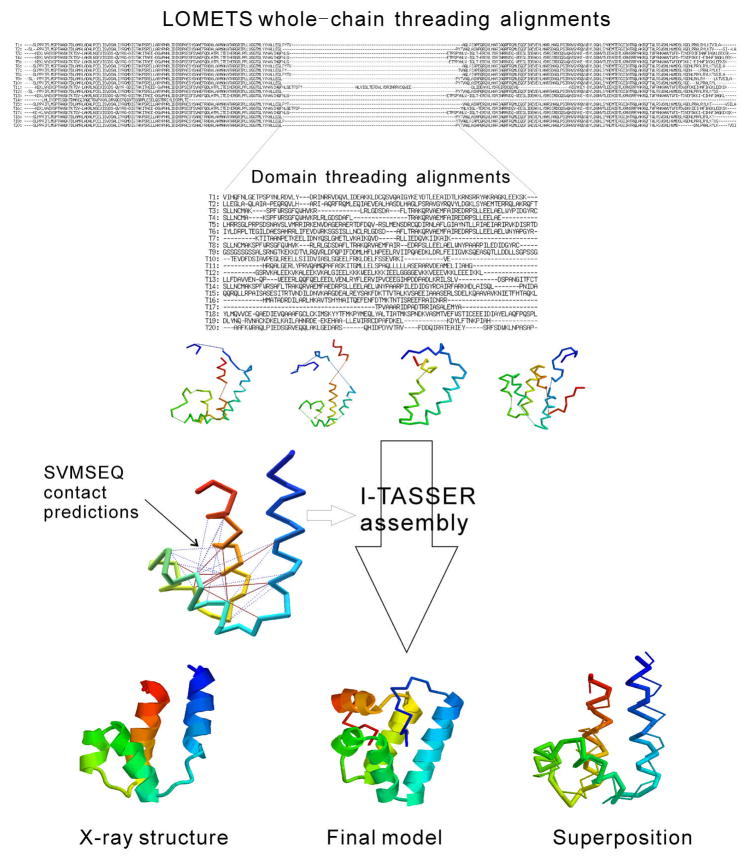
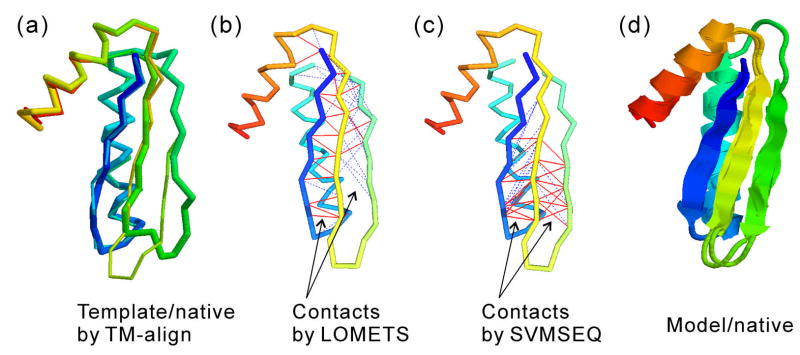
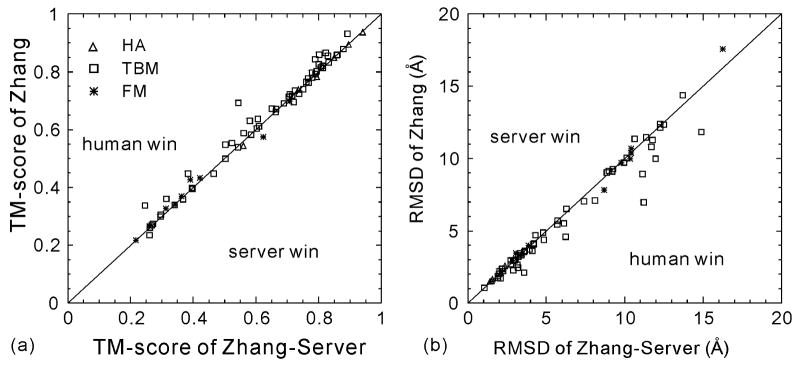
The I-TASSER algorithm for 3D protein structure prediction was tested in CASP8, with the procedure fully automated in both the Server and Human sections. The quality of the server models is close to that of human ones but the human predictions incorporate more diverse templates from other servers which improve the human predictions in some of the distant homology targets. For the first time, the sequence-based contact predictions from machine learning techniques are found helpful for both template-based modeling (TBM) and template-free modeling (FM). In TBM, although the accuracy of the sequence based contact predictions is on average lower than that from template-based ones, the novel contacts in the sequence-based predictions, which are complementary to the threading templates in the weakly or unaligned regions, are important to improve the global and local packing in these regions. Moreover, the newly developed atomic structural refinement algorithm was tested in CASP8 and found to improve the hydrogen-bonding networks and the overall TM-score, which is mainly due to its ability of removing steric clashes so that the models can be generated from cluster centroids. Nevertheless, one of the major issues of the I-TASSER pipeline is the model selection where the best models could not be appropriately recognized when the correct templates are detected only by the minority of the threading algorithms. There are also problems related with domain-splitting and mirror image recognition which mainly influences the performance of I-TASSER modeling in the FM-based structure predictions.
Copyright 2009 Wiley-Liss, Inc.
Figures
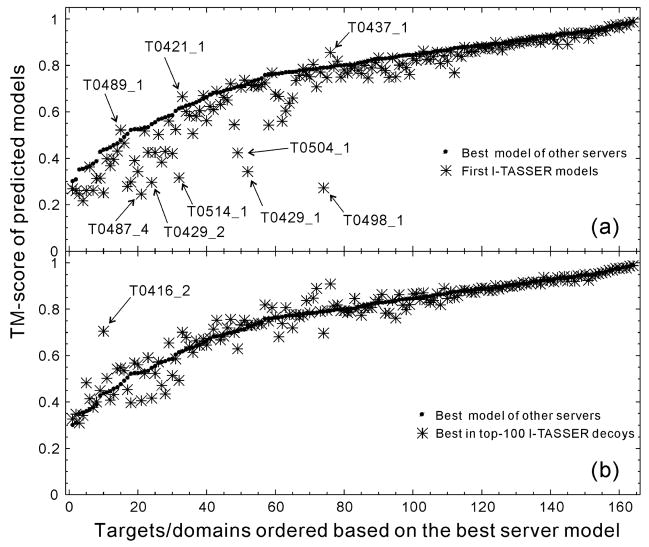
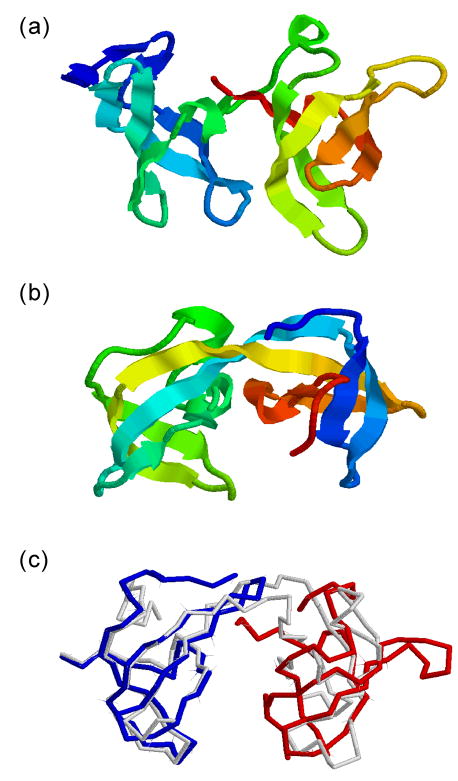
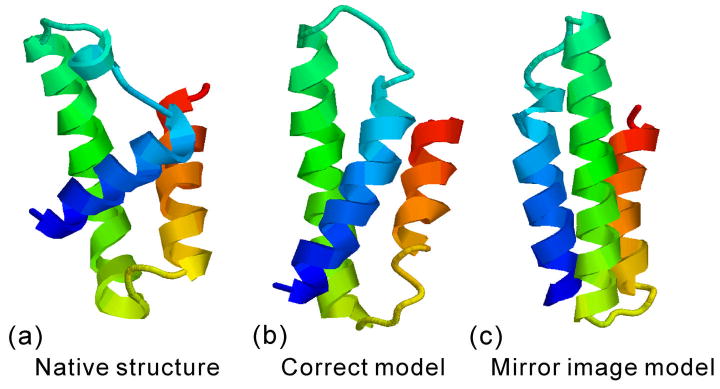
References
-
- Murzin AG, Bateman A. CASP2 knowledge-based approach to distant homology recognition and fold prediction in CASP4. Proteins. 2001;(Suppl 5):76–85. - PubMed
-
- Ginalski K, Rychlewski L. Protein structure prediction of CASP5 comparative modeling and fold recognition targets using consensus alignment approach and 3D assessment. Proteins. 2003;53 (Suppl 6):410–417. - PubMed
-
- Das R, Qian B, Raman S, Vernon R, Thompson J, Bradley P, Khare S, Tyka MD, Bhat D, Chivian D, Kim DE, Sheffler WH, Malmstrom L, Wollacott AM, Wang C, Andre I, Baker D. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins. 2007;69(S8):118–128. - PubMed
-
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294(5540):93–96. - PubMed
-
- Skolnick J, Fetrow JS, Kolinski A. Structural genomics and its importance for gene function analysis. Nat Biotechnol. 2000;18(3):283–287. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
