

Review
doi: 10.1021/cr9002193.
Hydrocarbon hydroxylation by cytochrome P450 enzymes
Affiliations
- PMID: 19769330
- PMCID: PMC2820140
- DOI: 10.1021/cr9002193
Item in Clipboard
Review
Hydrocarbon hydroxylation by cytochrome P450 enzymes
Chem Rev.
.
No abstract available
Figures

Summary of the cytochrome P450 catalytic cycle. The heme group is represented as two solid bars with the iron (Fe) between them. The cysteine thiolate provided by the protein is represented as an S. RH is a substrate hydrocarbon and ROH the resulting hydroxylated product. The +. over one of the heme bars indicates the radical cation is located on the porphyrins, whereas its placement besides the brackets indicates that the radical is located somewhere on the protein.

Comparison of the products from the oxidation of N-alkyl-N-cyclopropyl-p-chloroaniline by CYP2B1 supported by cytochrome P450 reductase, NADPH, and O2 with the primary products obtained when the oxidation is mediated by either horseradish peroxidase and H2O2 or by CYP2B1 and PhIO. The charged product shown in brackets is identified after it is trapped by cyanide ion.

Retention of stereochemistry in the hydroxylation of terminal methyl groups by cytochrome P450 enzymes.

The cytochrome P450-catalyzed hydroxylation of 2,3,5,6-tetradeuterated norbornane proceeds with partial invertion of stereochemistry at the carbon undergoing hydroxylation, implicating an intermediate such as a radical in the reaction mechanism.

Selected reactions in which deuterium isotope effects have been used to investigate the nature of cytochrome P450 catalysis.
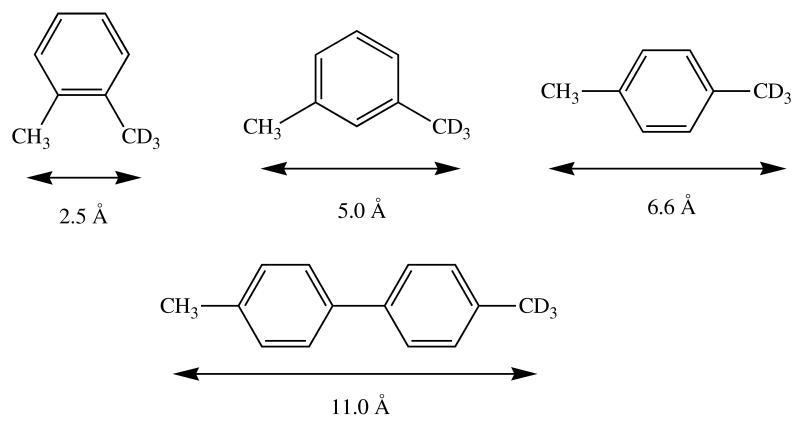
Low mobility of a substrate within the P450 active site can result in masking of the intrinsic isotope effect in a hydroxylation reaction. The isotope effects observed in the product distributions from hydroxylation of the CH3 versus CD3 methyl group in the compounds in this figure depend on the distance between the two groups on the substrate. The distance is indicated in the figure.
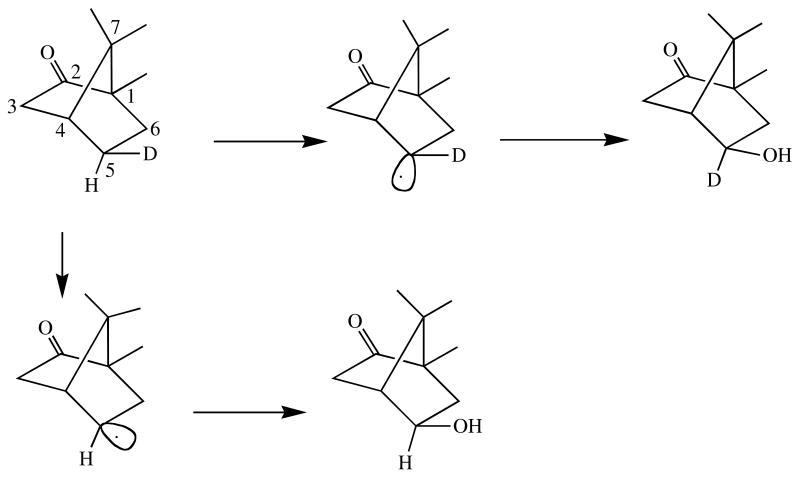
Inversion of stereochemistry in the hydroxylation of 5-exo-deuterated camphor by CYP101. The carbon atoms are numbered. Exo indicates the hydroxyl is on the same side of the molecule as the bridge with the highest numbered carbon.

Reactions in which the hydroxylation regiochemistry is altered due to shift of a double bond allylic to the site from which the hydrogen is removed.

Loss of stereochemistry observed in the hydroxylation of pulegone by a cytochrome P450 enzyme.
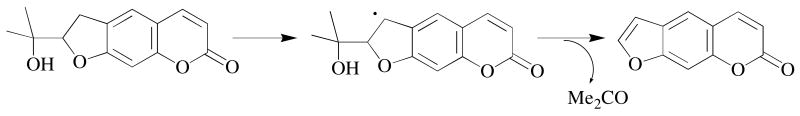
Cleavage of a carbon-carbon bond in the oxidation of marmesin by cytochrome P450 is best rationalized as occurring from the carbon radical generated by hydrogen abstraction from the dihydrofuran ring of the substrate.

Schematic of a radical clock reaction manifold. The rate kt can be estimated from the ratio of the unrearranged and rearranged products if the rate kr is known (assuming single state reactivity).

The oxidation of bicyclo[2.1.0]pentane, a radical clock substrate, by cytochrome P450 yields both a rearranged and an unrearranged hydroxylated product.
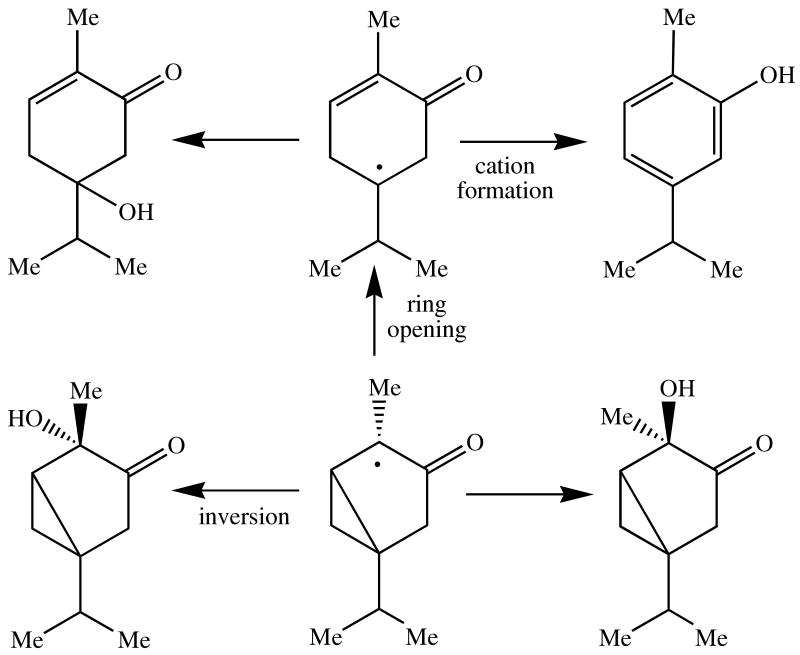
α- and β-Thujone are radical clocks whose C-4 radical can undergo two simultaneous, competing rearrangements, one a conventional cyclopropyl ring opening reaction and the other an inversion of the stereochemistry of a methyl group. The radical can also be converted to a cation which rearranges to a distinct product.

Rearrangements of a probe that gives different products depending on whether the intermediate is a radical or a cation.
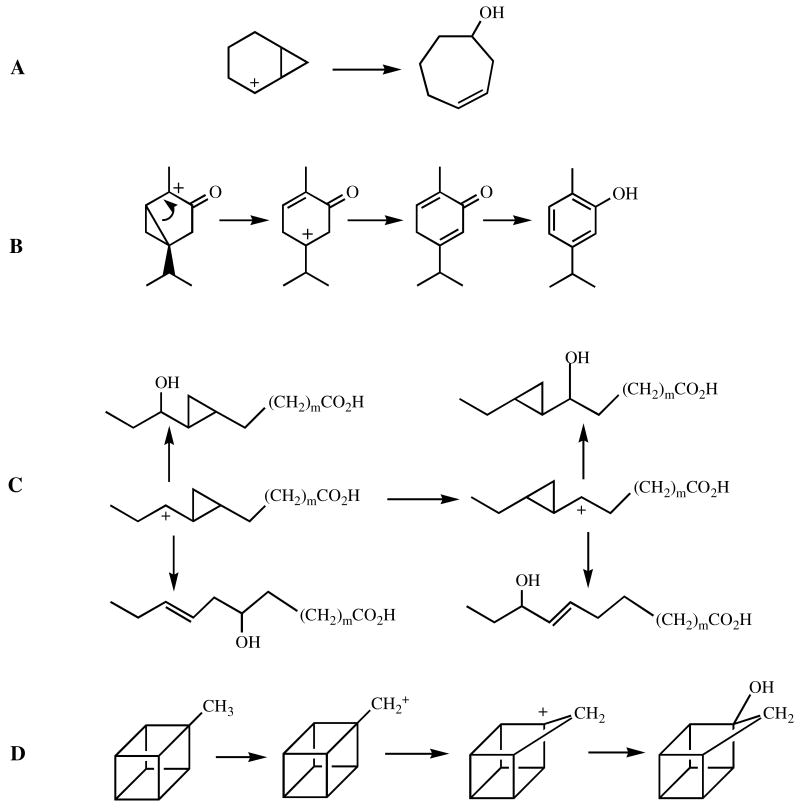
Rearrangements of various cations used to test for cation involvement in cytochrome P450 hydrocarbon oxidations.
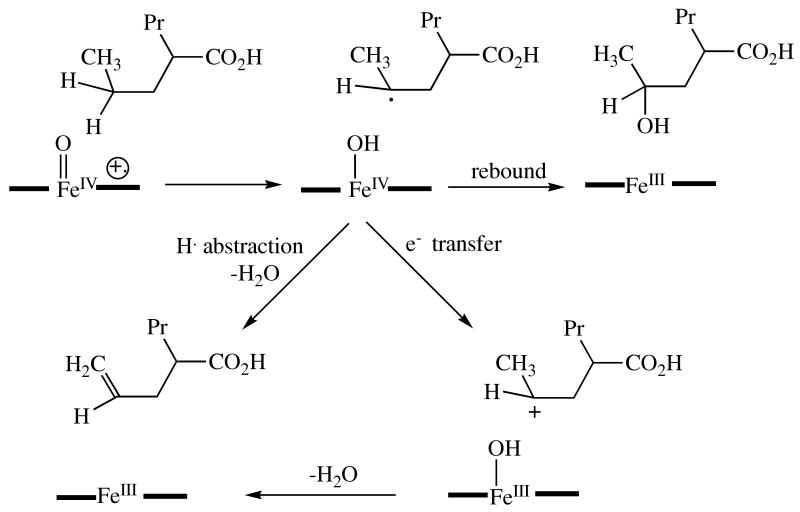
Desaturatoin of valproic acid via either a cation intermediate or two sequential hydrogen abstraction reactions. The heme is again represented as an iron atom between two black bars.
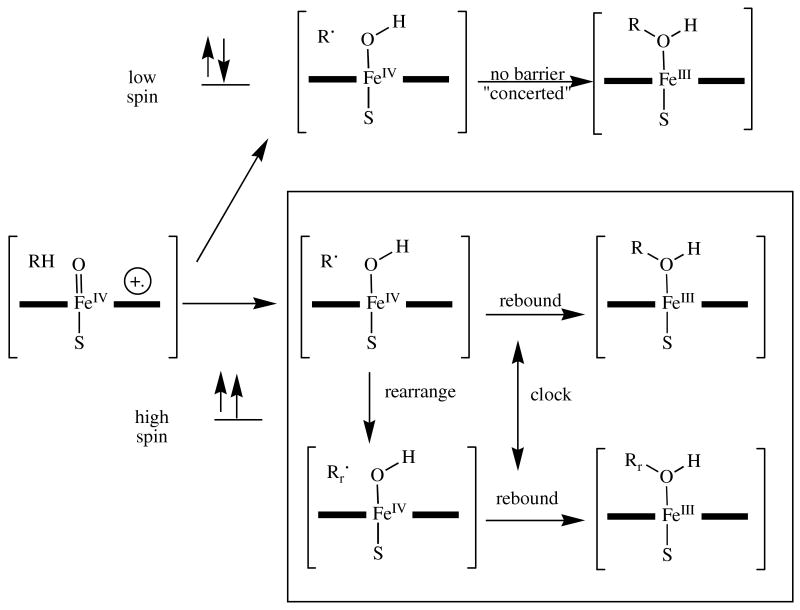
A schematic showing the rationalization for the observation of extremely fast radical recombination times with some radical clock substrates provided by the two-state hydroxylation hypothesis. The doublet (low) spin transition state collapses in a barrierless manner and is not able to undergo radical rearrangements, whereas the quartet (high) spin transition state (in the box) has a barrier to the recombination reaction that allows the radical rearrangement to occur. The radical clock thus only operates in the lower (high spin) trajectory, but the overall product ratios reflect the sum of the two pathways. As a result, the final product ratio has a deceptively large proportion of the unrearranged product.

Sites of hydroxylation of small hydrocarbons by liver microsomal cytochrome P450. The percent of hydroxylation at each site is shown. In the case of equivalent sites, the total hydroxylation at those sites has been divided equally among them.
References
-
- Guengerich FP. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. Ortiz de Montellano PR, editor. Kluwer Elsevier; New York: 2005. pp. 377–530.
-
- McLean KJ, Munro AW. Drug Metab Rev. 2008;40:427. - PubMed
-
- Ehlting J, Provart NJ, Werck-Reichhart D. Biochem Soc Trans. 2006;34:1192. - PubMed
-
- Schuler MA, Werck-Reichhart D. Annu Rev Plant Biol. 2003;54:629. - PubMed
-
- Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, Nebert DW. DNA Cell Biol. 1993;12:1. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
