

DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets
- PMID: 19775280
- PMCID: PMC2765589
- DOI: 10.1111/j.1476-5381.2009.00423.x
DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets
Abstract
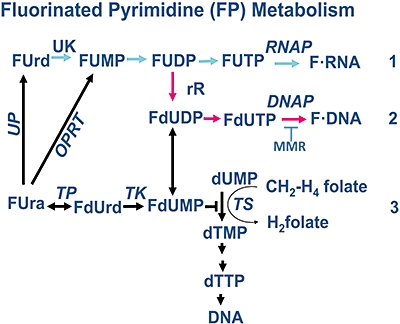
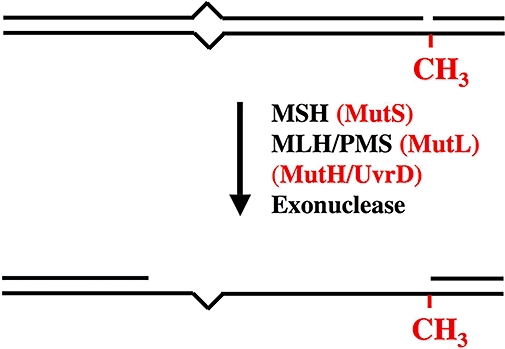
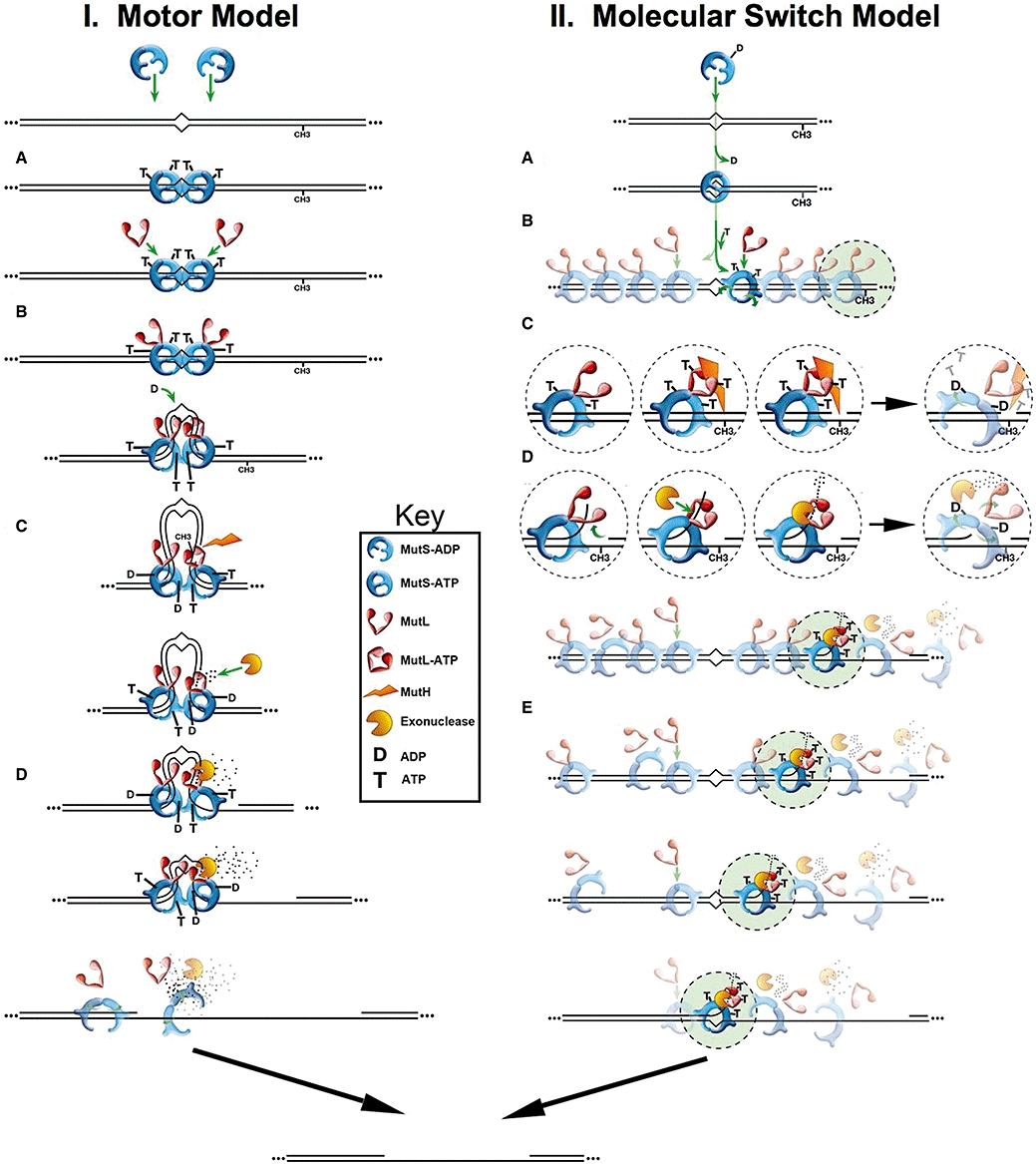
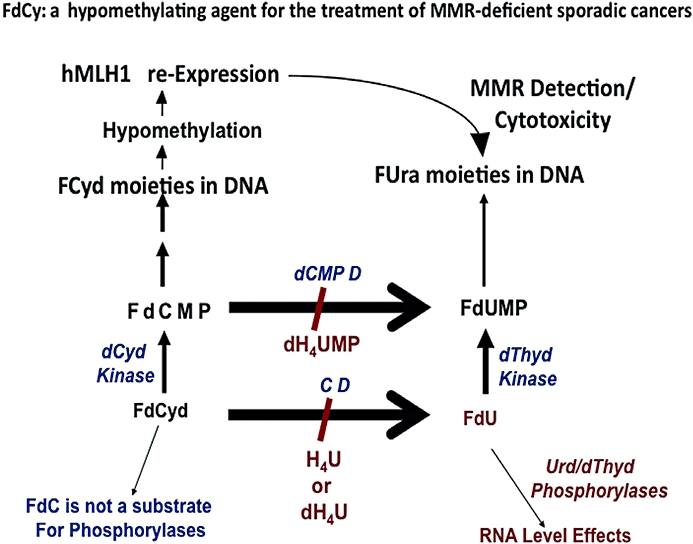
The metabolism and efficacy of 5-fluorouracil (FUra) and other fluorinated pyrimidine (FP) derivatives have been intensively investigated for over fifty years. FUra and its antimetabolites can be incorporated at RNA- and DNA-levels, with RNA level incorporation provoking toxic responses in human normal tissue, and DNA-level antimetabolite formation and incorporation believed primarily responsible for tumour-selective responses. Attempts to direct FUra into DNA-level antimetabolites, based on mechanism-of-action studies, have led to gradual improvements in tumour therapy. These include the use of leukovorin to stabilize the inhibitory thymidylate synthase-5-fluoro-2'-deoxyuridine 5' monophoshate (FdUMP)-5,10-methylene tetrahydrofolate (5,10-CH(2)FH(4)) trimeric complex. FUra incorporated into DNA also contributes to antitumour activity in preclinical and clinical studies. This review examines our current state of knowledge regarding the mechanistic aspects of FUra:Gua lesion detection by DNA mismatch repair (MMR) machinery that ultimately results in lethality. MMR-dependent direct cell death signalling or futile cycle responses will be discussed. As 10-30% of sporadic colon and endometrial tumours display MMR defects as a result of human MutL homologue-1 (hMLH1) promoter hypermethylation, we discuss the use and manipulation of the hypomethylating agent, 5-fluorodeoxycytidine (FdCyd), and our ability to manipulate its metabolism using the cytidine or deoxycytidylate (dCMP) deaminase inhibitors, tetrahydrouridine or deoxytetrahydrouridine, respectively, as a method for re-expression of hMLH1 and re-sensitization of tumours to FP therapy.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
