

Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants
- PMID: 19775921
- PMCID: PMC3721340
- DOI: 10.1016/j.ymgme.2009.08.003
Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants
Abstract
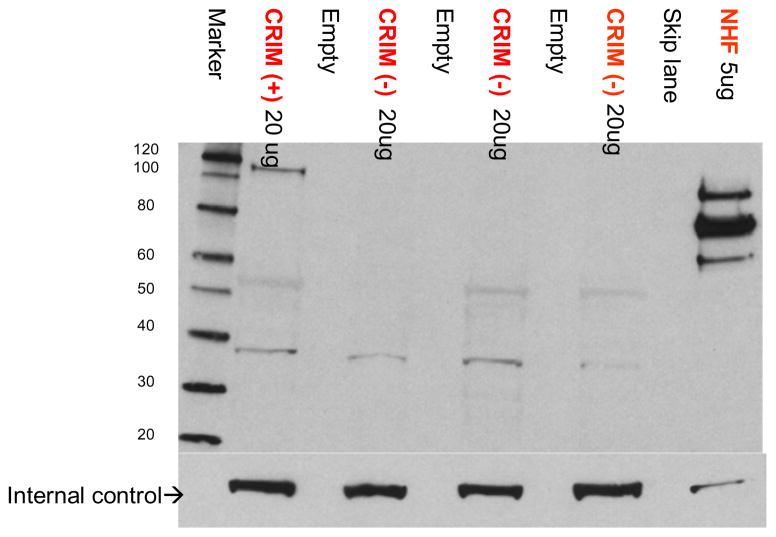
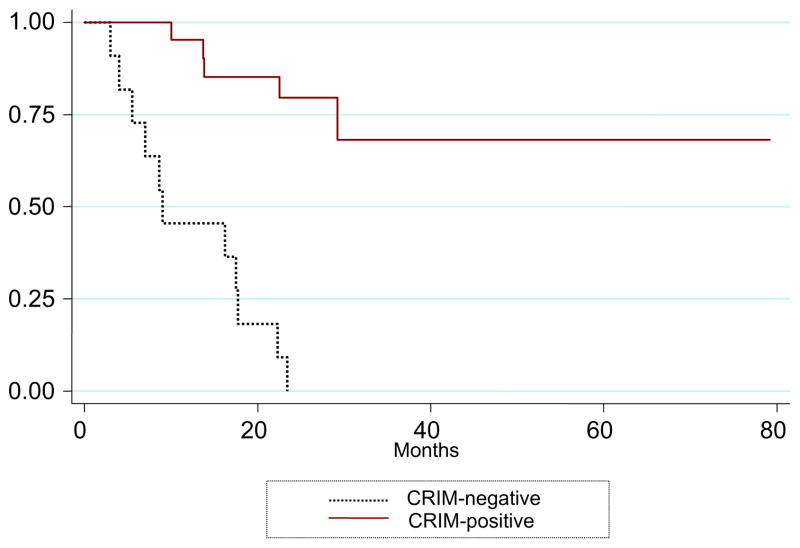
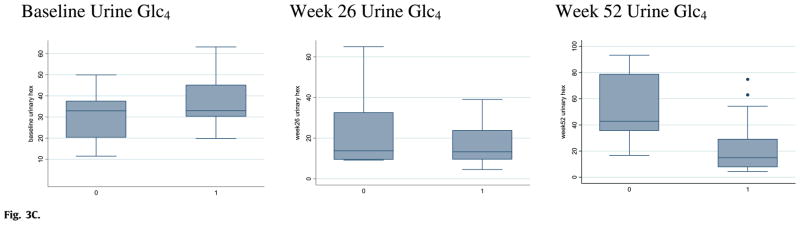
Deficiency of acid alpha glucosidase (GAA) causes Pompe disease, which is usually fatal if onset occurs in infancy. Patients synthesize a non-functional form of GAA or are unable to form native enzyme. Enzyme replacement therapy with recombinant human GAA (rhGAA) prolongs survival in infantile Pompe patients but may be less effective in cross-reactive immunologic material (CRIM)-negative patients. We retrospectively analyzed the influence of CRIM status on outcome in 21 CRIM-positive and 11 CRIM-negative infantile Pompe patients receiving rhGAA. Patients were from the clinical setting and from clinical trials of rhGAA, were 6 months of age, were not invasively ventilated, and were treated with IV rhGAA at a cumulative or total dose of 20 or 40 mg/kg/2 weeks. Outcome measures included survival, invasive ventilator-free survival, cardiac status, gross motor development, development of antibodies to rhGAA, and levels of urinary Glc(4). Following 52 weeks of treatment, 6/11 (54.5%) CRIM-negative and 1/21 (4.8%) CRIM-positive patients were deceased or invasively ventilated (p<0.0001). By age 27.1 months, all CRIM-negative patients and 4/21 (19.0%) CRIM-positive patients were deceased or invasively ventilated. Cardiac function and gross motor development improved significantly more in the CRIM-positive group. IgG antibodies to rhGAA developed earlier and serotiters were higher and more sustained in the CRIM-negative group. CRIM-negative status predicted reduced overall survival and invasive ventilator-free survival and poorer clinical outcomes in infants with Pompe disease treated with rhGAA. The effect of CRIM status on outcome appears to be mediated by antibody responses to the exogenous protein.
Trial registration: ClinicalTrials.gov NCT00025896 NCT00053573 NCT00059280 NCT00074919 NCT00125879.
Figures
References
-
- Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. - PubMed
-
- van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. - PubMed
-
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, Sandkuijl LA, Reuser AJ, van der Ploeg AT. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–716. - PubMed
-
- Chien YH, Chiang SC, Zhang XK, Keutzer J, Lee NC, Huang AC, Chen CA, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Early detection of Pompe Disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics. 2008 - PubMed
-
- Fuller M, Van der Ploeg A, Reuser AJ, Anson DS, Hopwood JJ. Isolation and characterisation of a recombinant, precursor form of lysosomal acid alpha-glucosidase. Eur J Biochem. 1995;234:903–909. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
