

Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns
- PMID: 19776032
- PMCID: PMC4481584
- DOI: 10.1093/hmg/ddp445
Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns
Abstract
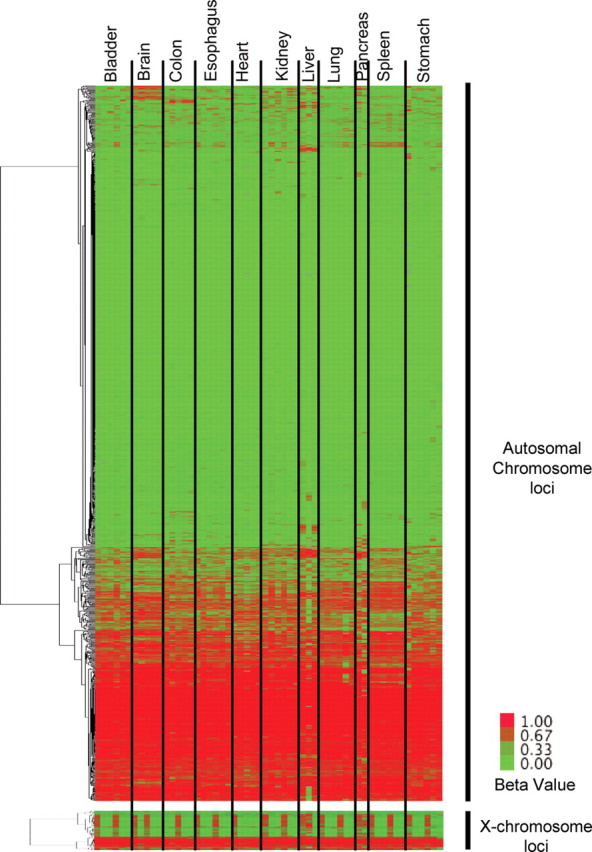
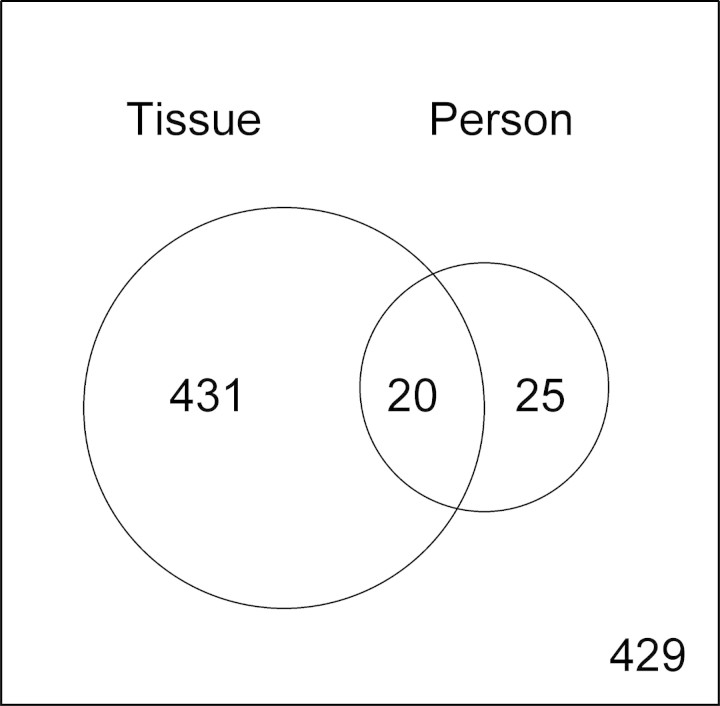
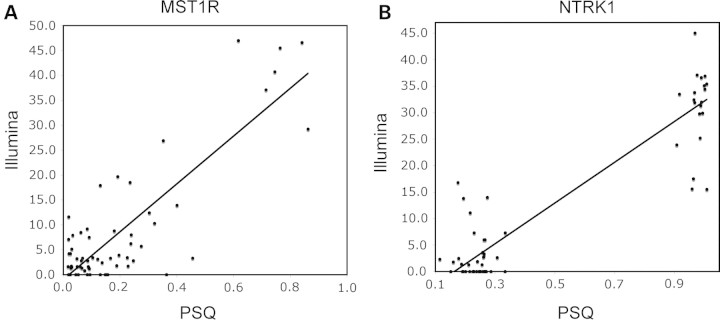
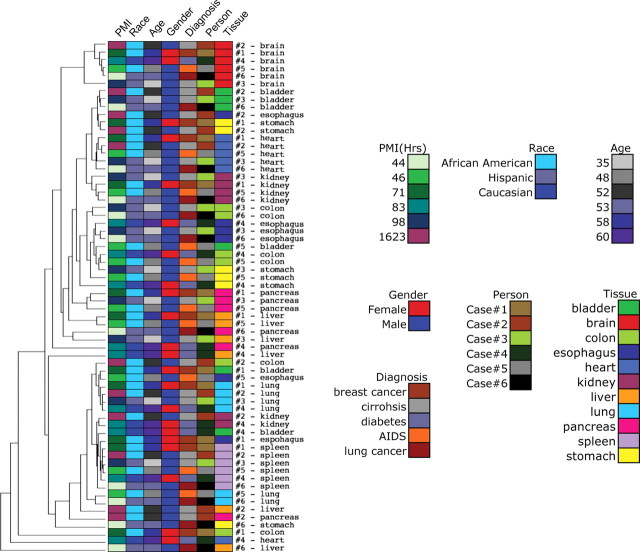
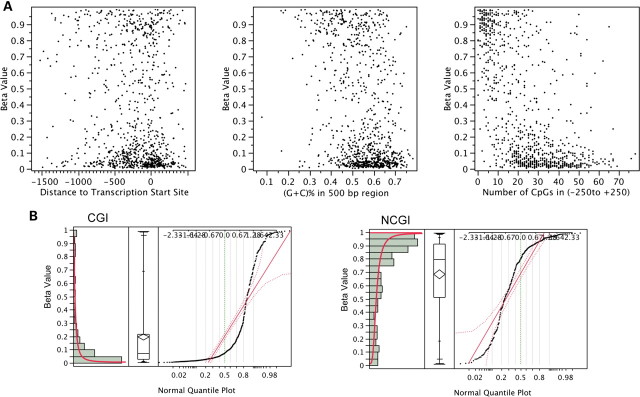
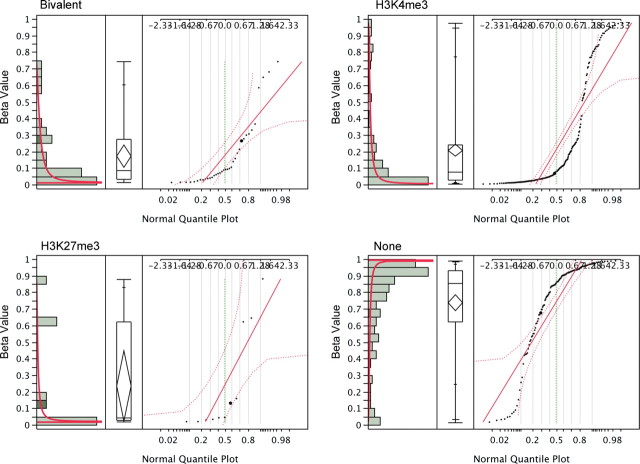
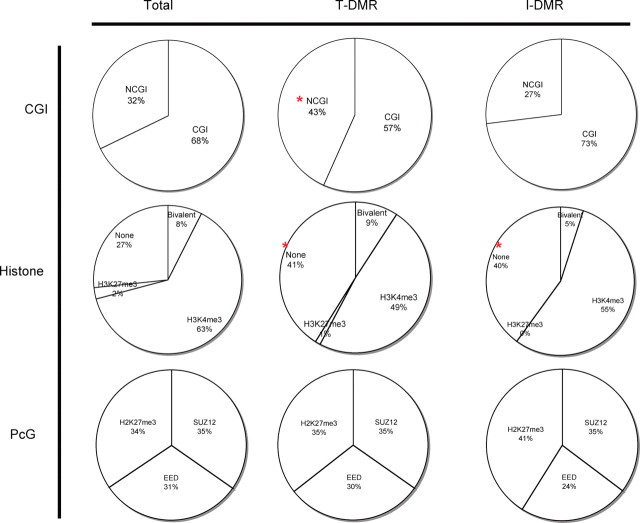
DNA methylation is known to be associated with cell differentiation, aging, disease and cancer. There exists an expanding base of knowledge regarding tissue-specific DNA methylation, but we have little information about person-specific DNA methylation. Here, we analyze the DNA methylation patterns of multiple tissues from multiple individuals using a high-throughput quantitative assay of genome-wide DNA methylation, namely the Illumina GoldenGate BeadArray. DNA methylation patterns were largely conserved across 11 different tissues (r = 0.852) and across six individuals (r = 0.829), and we found that DNA was highly methylated in non-CpG islands and/or CpG sites that are not occupied by either H3K4me3 or H3K27me3 (P < 0.05). Finally, we found that the Illumina GoldenGate assay features a large number of probes (265/1505 probes, 17.6%) that contain single-nucleotide polymorphisms, which may interfere with DNA methylation analyses in genome-wide studies.
Figures
References
-
- Bibikova M., Laurent L.C., Ren B., Loring J.F., Fan J.B. Unraveling epigenetic regulation in embryonic stem cells. Cell Stem Cell. 2008;2:123–134. - PubMed
-
- Hellman A., Chess A. Gene body-specific methylation on the active X-chromosome. Science (New York, N.Y.) 2007;315:1141–1143. - PubMed
-
- Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. - PubMed
-
- Richardson B.C. Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J. Nutr. 2002;132:2401S–2405S. - PubMed
-
- Ahuja N., Li Q., Mohan A.L., Baylin S.B., Issa J.P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
