

High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans
- PMID: 19778439
- PMCID: PMC2768976
- DOI: 10.1186/gb-2009-10-9-r101
High resolution transcriptome maps for wild-type and nonsense-mediated decay-defective Caenorhabditis elegans
Abstract
Background: While many genome sequences are complete, transcriptomes are less well characterized. We used both genome-scale tiling arrays and massively parallel sequencing to map the Caenorhabditis elegans transcriptome across development. We utilized this framework to identify transcriptome changes in animals lacking the nonsense-mediated decay (NMD) pathway.
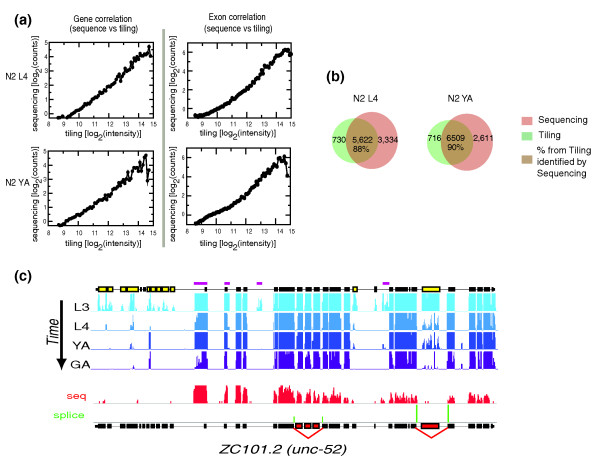
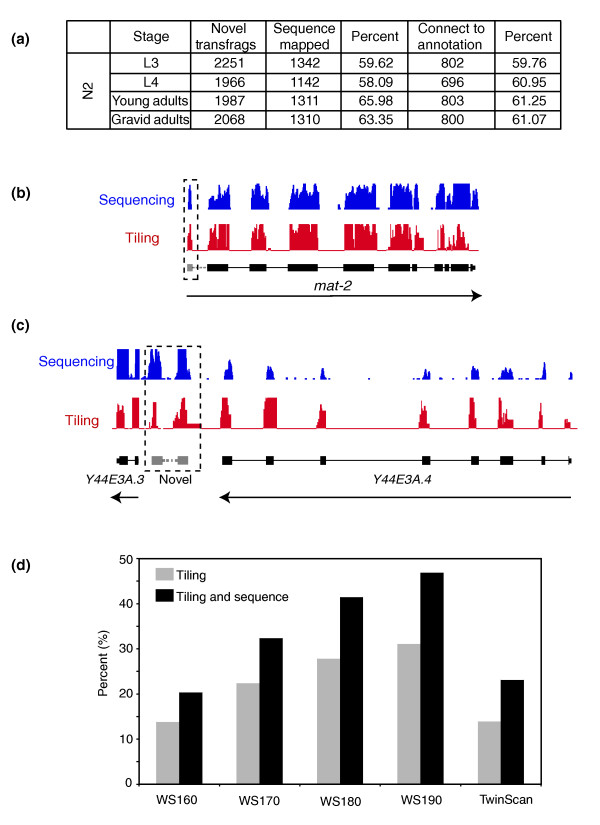
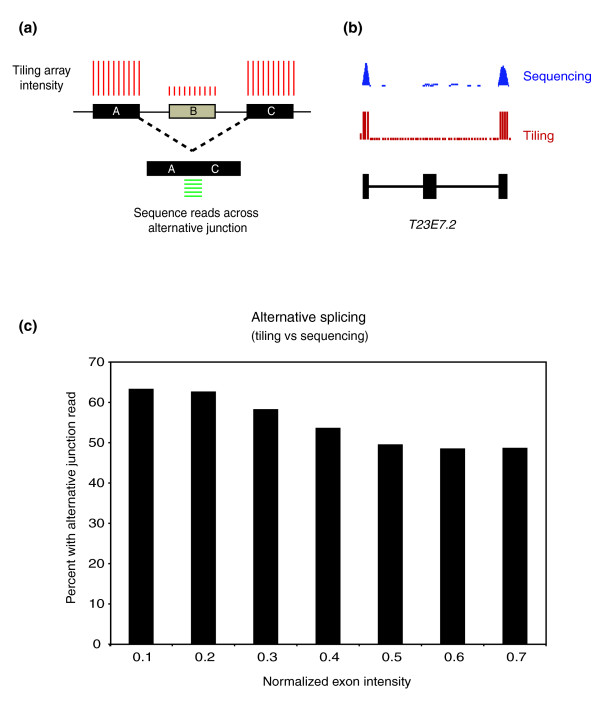
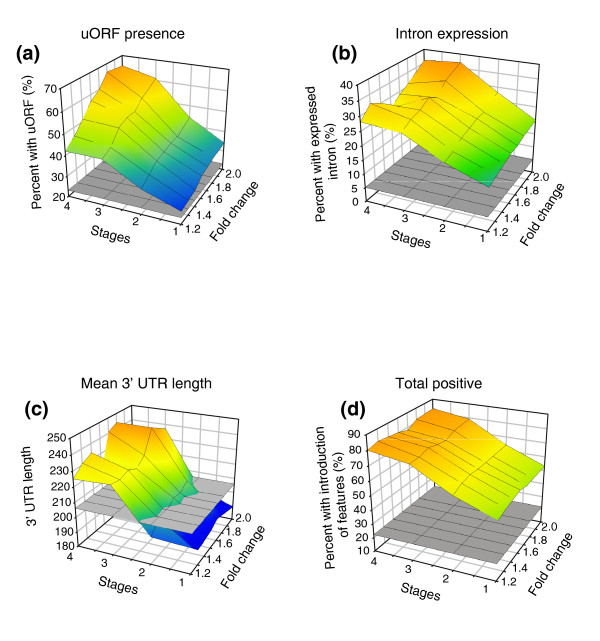
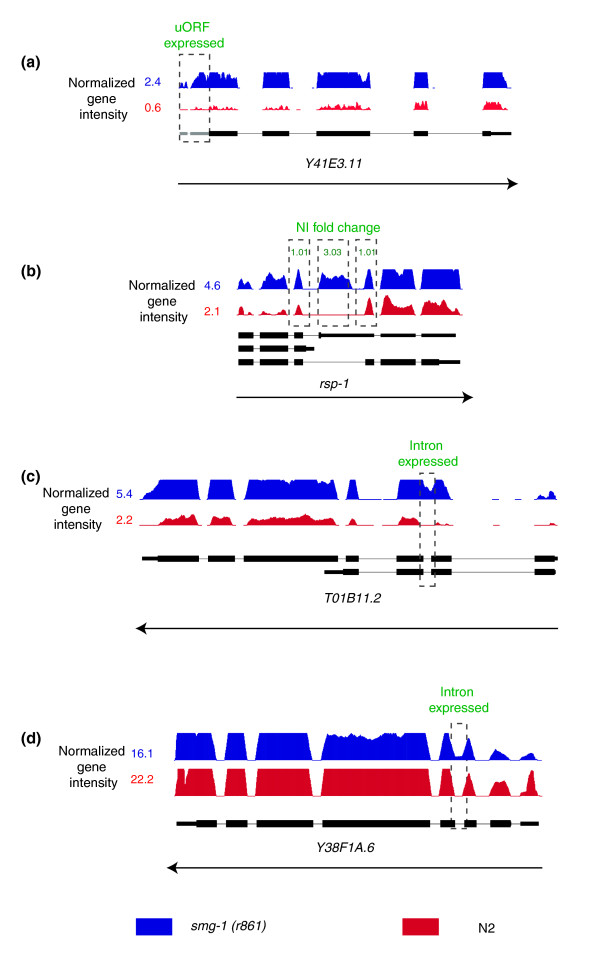
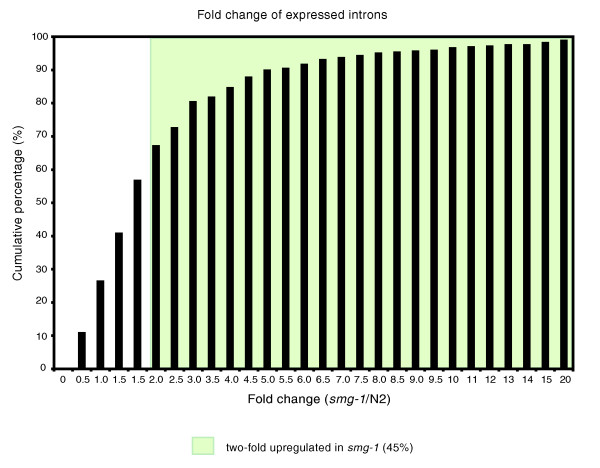
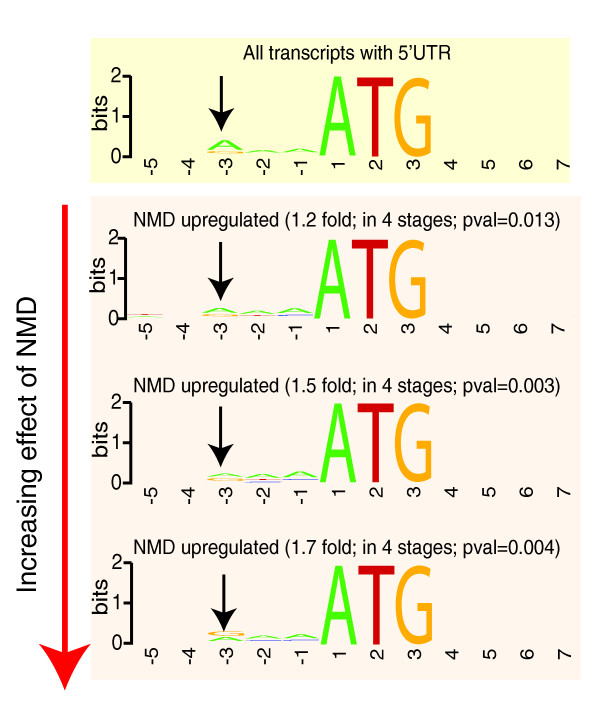
Results: We find that while the majority of detectable transcripts map to known gene structures, >5% of transcribed regions fall outside current gene annotations. We show that >40% of these are novel exons. Using both technologies to assess isoform complexity, we estimate that >17% of genes change isoform across development. Next we examined how the transcriptome is perturbed in animals lacking NMD. NMD prevents expression of truncated proteins by degrading transcripts containing premature termination codons. We find that approximately 20% of genes produce transcripts that appear to be NMD targets. While most of these arise from splicing errors, NMD targets are enriched for transcripts containing open reading frames upstream of the predicted translational start (uORFs). We identify a relationship between the Kozak consensus surrounding the true start codon and the degree to which uORF-containing transcripts are targeted by NMD and speculate that translational efficiency may be coupled to transcript turnover via the NMD pathway for some transcripts.
Conclusions: We generated a high-resolution transcriptome map for C. elegans and used it to identify endogenous targets of NMD. We find that these transcripts arise principally through splicing errors, strengthening the prevailing view that splicing and NMD are highly interlinked processes.
Figures
Similar articles
-
The Substrates of Nonsense-Mediated mRNA Decay in Caenorhabditis elegans.G3 (Bethesda). 2018 Jan 4;8(1):195-205. doi: 10.1534/g3.117.300254. G3 (Bethesda). 2018. PMID: 29122854 Free PMC article.
-
Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans.RNA. 2009 Sep;15(9):1652-60. doi: 10.1261/rna.1711109. Epub 2009 Jul 17. RNA. 2009. PMID: 19617316 Free PMC article.
-
Multiple transcripts from a 3'-UTR reporter vary in sensitivity to nonsense-mediated mRNA decay in Saccharomyces cerevisiae.PLoS One. 2013 Nov 18;8(11):e80981. doi: 10.1371/journal.pone.0080981. eCollection 2013. PLoS One. 2013. PMID: 24260526 Free PMC article.
-
The Branched Nature of the Nonsense-Mediated mRNA Decay Pathway.Trends Genet. 2021 Feb;37(2):143-159. doi: 10.1016/j.tig.2020.08.010. Epub 2020 Sep 29. Trends Genet. 2021. PMID: 33008628 Free PMC article. Review.
-
[Nonsense-mediated mRNA decay (NMD)--on guard of mRNA quality].Postepy Biochem. 2006;52(4):390-8. Postepy Biochem. 2006. PMID: 17536508 Review. Polish.
Cited by
-
Nutritional control of mRNA isoform expression during developmental arrest and recovery in C. elegans.Genome Res. 2012 Oct;22(10):1920-9. doi: 10.1101/gr.133587.111. Epub 2012 Apr 26. Genome Res. 2012. PMID: 22539650 Free PMC article.
-
Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability.RNA Biol. 2012 Nov;9(11):1370-9. doi: 10.4161/rna.22360. Epub 2012 Oct 12. RNA Biol. 2012. PMID: 23064114 Free PMC article.
-
Defining nonsense-mediated mRNA decay intermediates in human cells.Methods. 2019 Feb 15;155:68-76. doi: 10.1016/j.ymeth.2018.12.005. Epub 2018 Dec 19. Methods. 2019. PMID: 30576707 Free PMC article. Review.
-
uORFdb--a comprehensive literature database on eukaryotic uORF biology.Nucleic Acids Res. 2014 Jan;42(Database issue):D60-7. doi: 10.1093/nar/gkt952. Epub 2013 Oct 24. Nucleic Acids Res. 2014. PMID: 24163100 Free PMC article.
-
The loss of SMG1 causes defects in quality control pathways in Physcomitrella patens.Nucleic Acids Res. 2018 Jun 20;46(11):5822-5836. doi: 10.1093/nar/gky225. Nucleic Acids Res. 2018. PMID: 29596649 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
