

Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects
- PMID: 19781108
- PMCID: PMC3279956
- DOI: 10.1017/S1462399409001203
Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects
Abstract
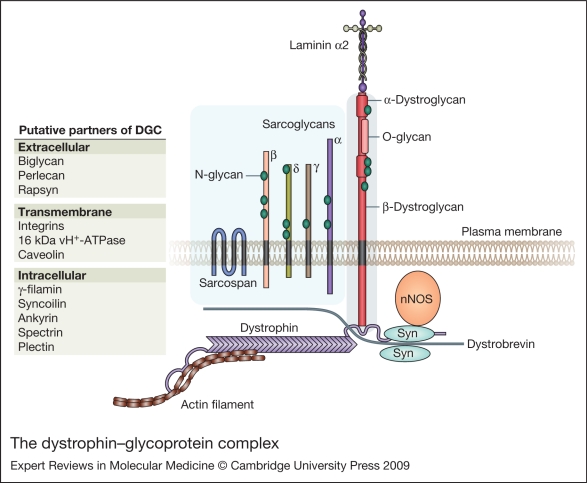
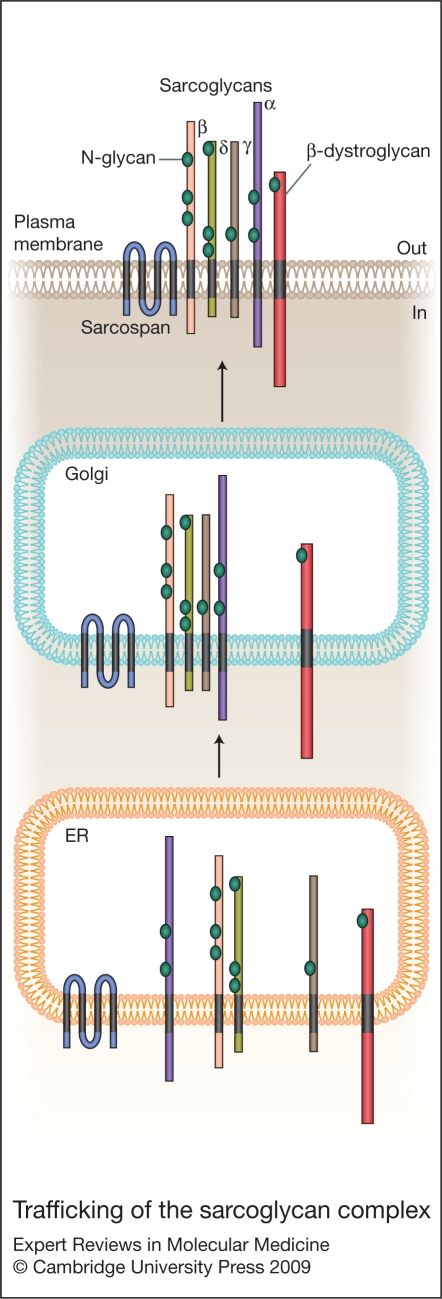
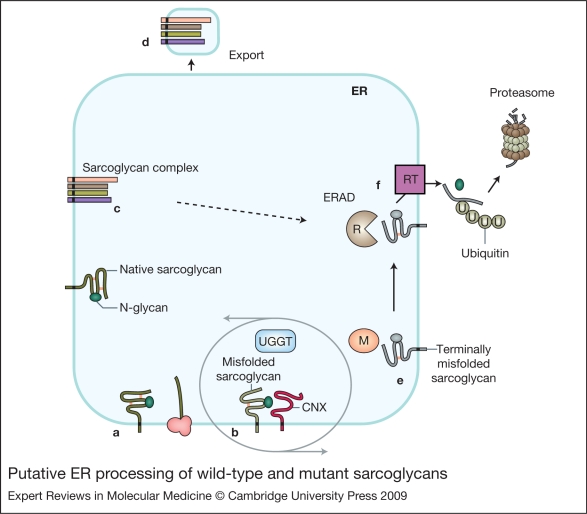
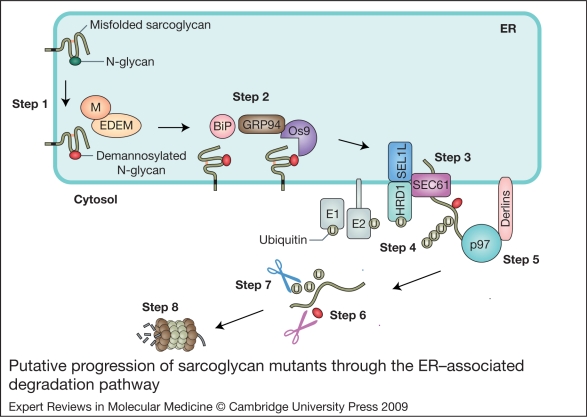
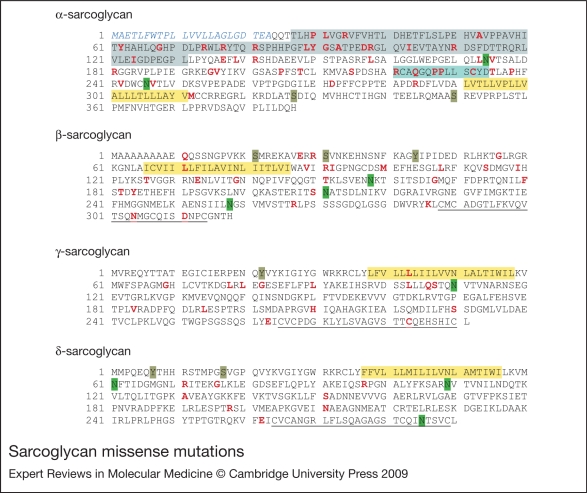
Sarcoglycanopathies are a group of autosomal recessive muscle-wasting disorders caused by genetic defects in one of four cell membrane glycoproteins, alpha-, beta-, gamma- or delta-sarcoglycan. These four sarcoglycans form a subcomplex that is closely linked to the major dystrophin-associated protein complex, which is essential for membrane integrity during muscle contraction and provides a scaffold for important signalling molecules. Proper assembly, trafficking and targeting of the sarcoglycan complex is of vital importance, and mutations that severely perturb tetramer formation and localisation result in sarcoglycanopathy. Gene defects in one sarcoglycan cause the absence or reduced concentration of the other subunits. Most genetic defects generate mutated proteins that are degraded through the cell's quality control system; however, in many cases, conformational modifications do not affect the function of the protein, yet it is recognised as misfolded and prematurely degraded. Recent evidence shows that misfolded sarcoglycans could be rescued to the cell membrane by assisting their maturation along the ER secretory pathway. This review summarises the etiopathogenesis of sarcoglycanopathies and highlights the quality control machinery as a potential pharmacological target for therapy of these genetic disorders.
Figures
References
-
- Blake D.J.. et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiological Reviews. 2002;82:291–329. - PubMed
-
- Davies K.E., Nowak K.J.. Molecular mechanisms of muscular dystrophies: old and new players. Nature Reviews. Molecular Cell Biology. 2006;7:762–773. - PubMed
-
- Hack A.A.. et al. Sarcoglycans in muscular dystrophy. Microscopy Research and Technique. 2000;48:167–180. - PubMed
-
- Ozawa E.. et al. Molecular and cell biology of the sarcoglycan complex. Muscle and Nerve. 2005;32:563–576. - PubMed
-
- Bönnemann C.G.. et al. β-Sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nature Genetics. 1995;11:266–273. - PubMed
Further reading, resources and contacts
Publications
-
- Muir L.A., Chamberlain J.S.. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Reviews in Molecular Medicine. 2009;11:e8. - PMC - PubMed
-
This review describes novel therapeutical interventions developed to cure Duchenne muscular dystrophy and that can be extended to other dystrophies.
-
- Linde L., Kerem B.. Introducing sense into nonsense in treatments of human genetic diseases. Trends in Genetics. 2008;24:552–563. - PubMed
-
This review deals with therapeutical approaches aimed at curing nonsense mutations creating a premature stop codon.
-
- Braun S.. Muscular gene transfer using nonviral vectors. Current Gene Therapy. 2008;8:391–405. - PubMed
-
This review reports on recent progresses of plasmid delivery of genes to muscles by using non-viral vectors.
Websites
-
- http://www.dmd.nl/sgca_seqvar.html http://www.dmd.nl/sgca_seqvar.html
-
The Muscular Dystrophy Association, MDA, together with numerous international associates, provides information on muscular dystrophies and helpful resources for patients:
-
- http://www.mda.org/ http://www.mda.org/
-
Treat Neuromuscular Disorders (TREAT-NMD) provide ample information about muscular dystrophies and updates on new therapeutic developments:
-
- http://www.treat-nmd.eu/home.php http://www.treat-nmd.eu/home.php
-
The Center for Neuromuscular Diseases, CNMD, is dedicated to clinical trials and therapy:
-
- http://www.cnmd.ac.uk/index.html http://www.cnmd.ac.uk/index.html
-
ClinicalTrialsGov offers a complete list of worldwide clinical trials on muscular dystrophies, and other medical conditions:
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
