

Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories
- PMID: 19783593
- PMCID: PMC2877550
- DOI: 10.1093/molbev/msp228
Rapid likelihood analysis on large phylogenies using partial sampling of substitution histories
Abstract
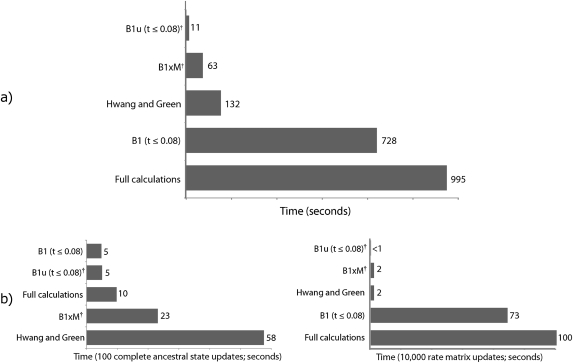
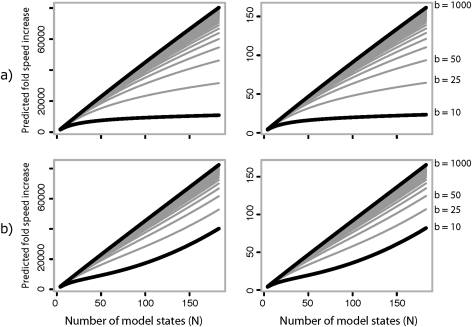
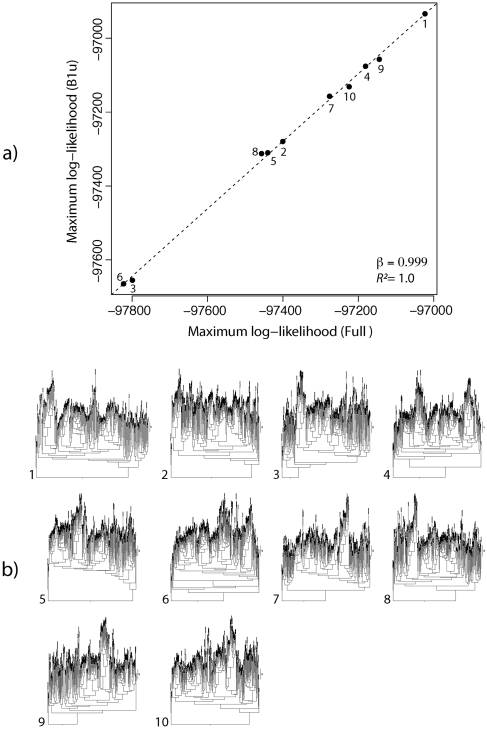
Likelihood-based approaches can reconstruct evolutionary processes in greater detail and with better precision from larger data sets. The extremely large comparative genomic data sets that are now being generated thus create new opportunities for understanding molecular evolution, but analysis of such large quantities of data poses escalating computational challenges. Recently developed Markov chain Monte Carlo methods that augment substitution histories are a promising approach to alleviate these computational costs. We analyzed the computational costs of several such approaches, considering how they scale with model and data set complexity. This provided a theoretical framework to understand the most important computational bottlenecks, leading us to combine novel variations of our conditional pathway integration approach with recent advances made by others. The resulting technique ("partial sampling" of substitution histories) is considerably faster than all other approaches we considered. It is accurate, simple to implement, and scales exceptionally well with dimensions of model complexity and data set size. In particular, the time complexity of sampling unobserved substitution histories using the new method is much faster than previously existing methods, and model parameter and branch length updates are independent of data set size. We compared the performance of methods on a 224-taxon set of mammalian cytochrome-b sequences. For a simple nucleotide substitution model, partial sampling was at least 10 times faster than the PhyloBayes program, which samples substitutions in continuous time, and about 100 times faster than when using fully integrated substitution histories. Under a general reversible model of amino acid substitution, the partial sampling method was 1,600 times faster than when using fully integrated substitution histories, confirming significantly improved scaling with model state-space complexity. Partial sampling of substitutions thus dramatically improves the utility of likelihood approaches for analyzing complex evolutionary processes on large data sets.
Figures
References
-
- Cormen TH. Introduction to algorithms. Cambridge (MA): The MIT Press; 2009.
-
- de Koning APJ, Gu W, Pollock DD. Ancestral sequence reconstruction in primate mitochondrial DNA: compositional bias and effect on functional inference (erratum) Mol Biol Evol. 2009;26:481. - PubMed
-
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. - PubMed
-
- Gelman A, Rubin DB, Carlin JB, Stern HS. Bayesian data analysis. London, New York: Chapman and Hall; 1992.
-
- Geman S, Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans Pattern Anal Machine Intel. 1984;6:721–741. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
