

Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension
- PMID: 19785764
- PMCID: PMC2762975
- DOI: 10.1186/1465-9921-10-87
Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension
Abstract
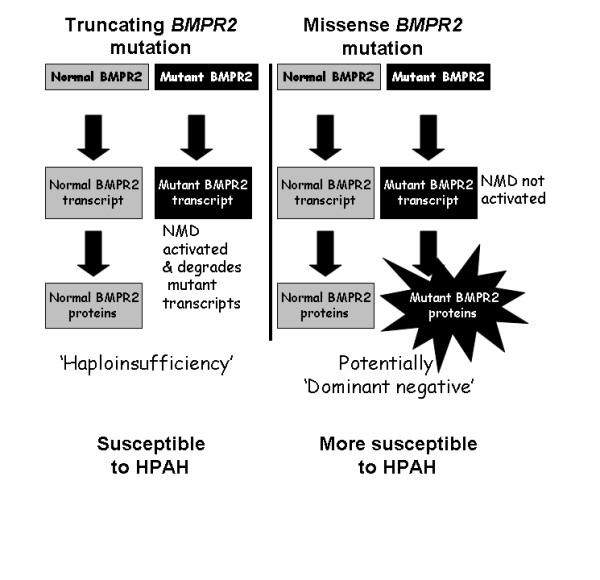
Background: Autosomal dominant inheritance of germline mutations in the bone morphogenetic protein receptor type 2 (BMPR2) gene are a major risk factor for pulmonary arterial hypertension (PAH). While previous studies demonstrated a difference in severity between BMPR2 mutation carriers and noncarriers, it is likely disease severity is not equal among BMPR2 mutations. We hypothesized that patients with missense BMPR2 mutations have more severe disease than those with truncating mutations.
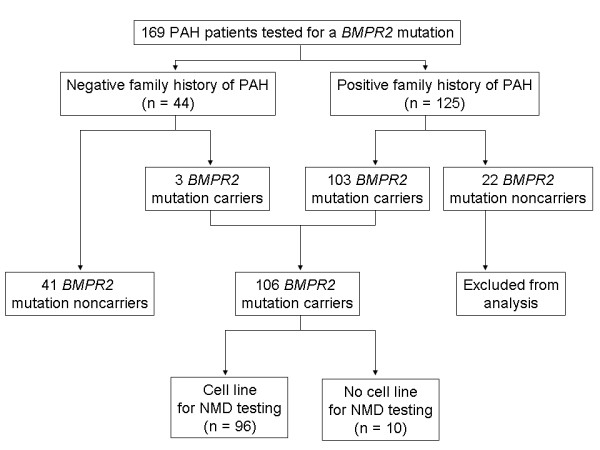
Methods: Testing for BMPR2 mutations was performed in 169 patients with PAH (125 with a family history of PAH and 44 with sporadic disease). Of the 106 patients with a detectable BMPR2 mutation, lymphocytes were available in 96 to functionally assess the nonsense-mediated decay pathway of RNA surveillance. Phenotypic characteristics were compared between BMPR2 mutation carriers and noncarriers, as well as between those carriers with a missense versus truncating mutation.
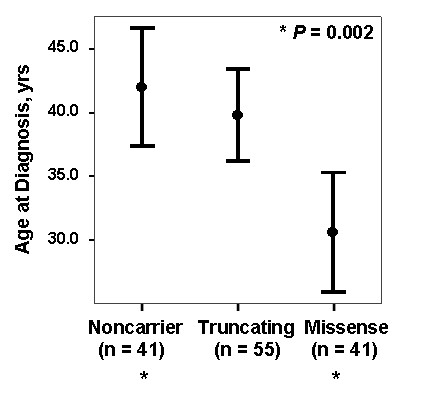
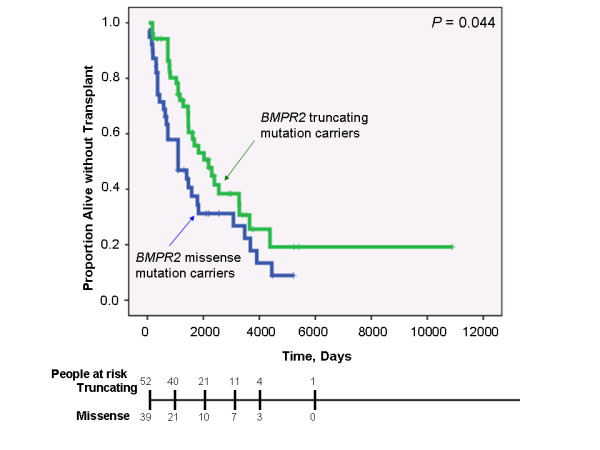
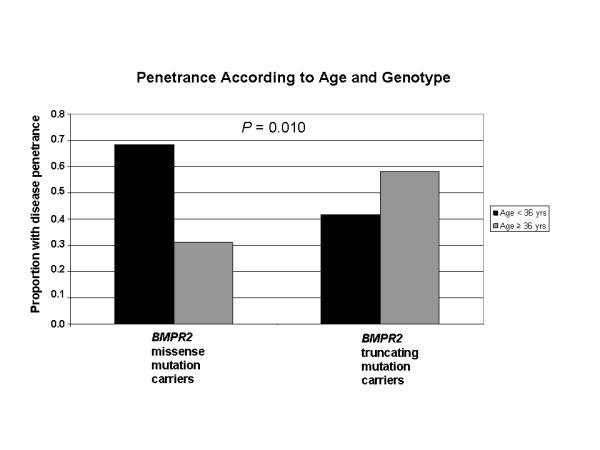
Results: While there was a statistically significant difference in age at diagnosis between carriers and noncarriers, subgroup analysis revealed this to be the case only for females. Among carriers, there was no difference in age at diagnosis, death, or survival according to exonic location of the BMPR2 mutation. However, patients with missense mutations had statistically significant younger ages at diagnosis and death, as well as shorter survival from diagnosis to death or lung transplantation than those with truncating mutations. Consistent with this data, the majority of missense mutations were penetrant prior to age 36 years, while the majority of truncating mutations were penetrant after age 36 years.
Conclusion: In this cohort, BMPR2 mutation carriers have more severe PAH disease than noncarriers, but this is only the case for females. Among carriers, patients with missense mutations that escape nonsense-mediated decay have more severe disease than those with truncating mutations. These findings suggest that treatment and prevention strategies directed specifically at BMPR2 pathway defects may need to vary according to the type of mutation.
Figures
References
-
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ. et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. doi: 10.1086/303059. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
