

Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis
- PMID: 19786028
- PMCID: PMC2813409
- DOI: 10.1053/j.gastro.2009.09.045
Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis
Abstract
Background & aims: Necrotizing enterocolitis (NEC), the leading cause of gastrointestinal death from gastrointestinal disease in preterm infants, is characterized by exaggerated TLR4 signaling and decreased enterocyte proliferation through unknown mechanisms. Given the importance of beta-catenin in regulating proliferation of many cell types, we hypothesize that TLR4 impairs enterocyte proliferation in NEC via impaired beta-catenin signaling.
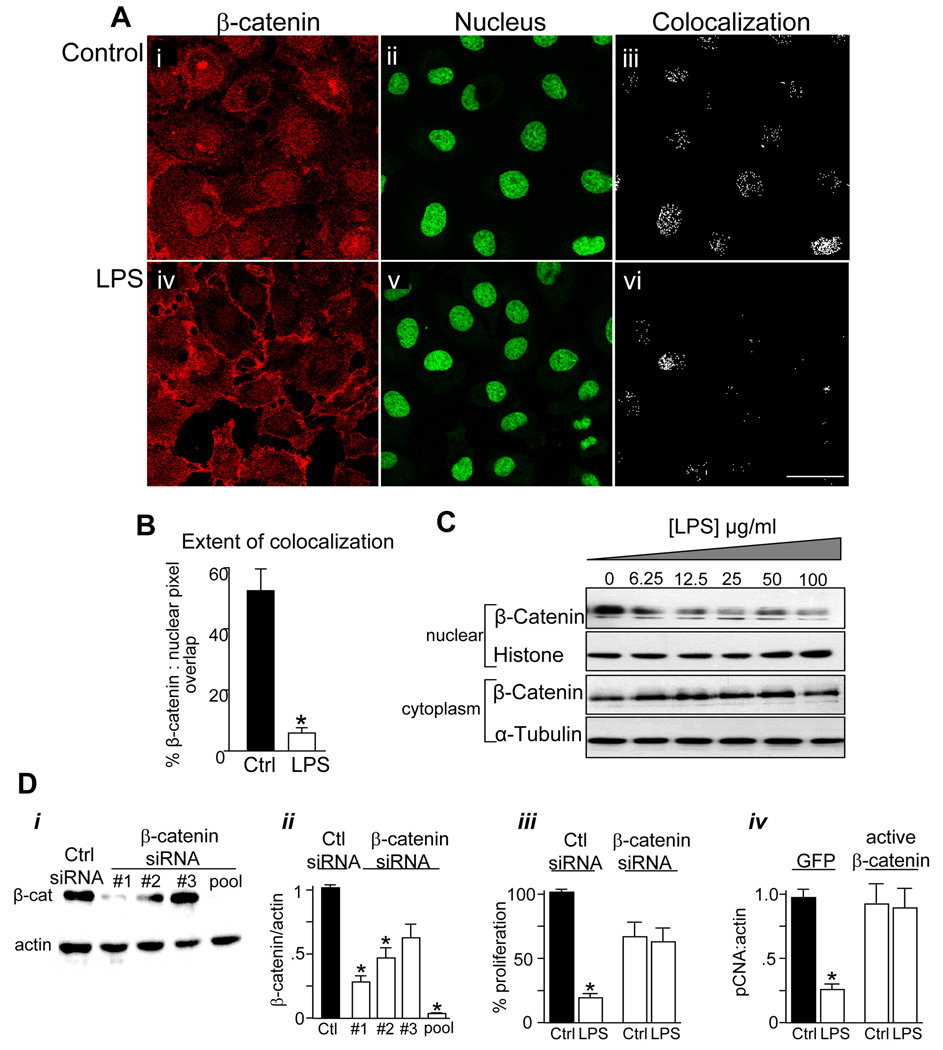
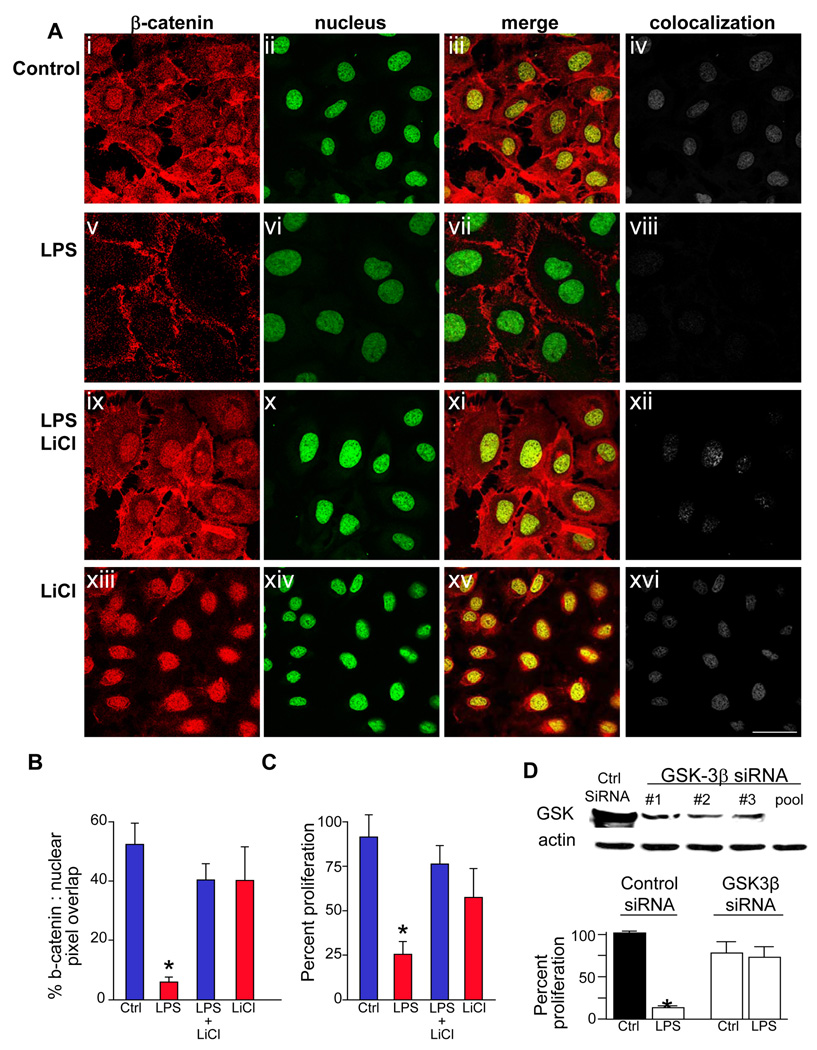
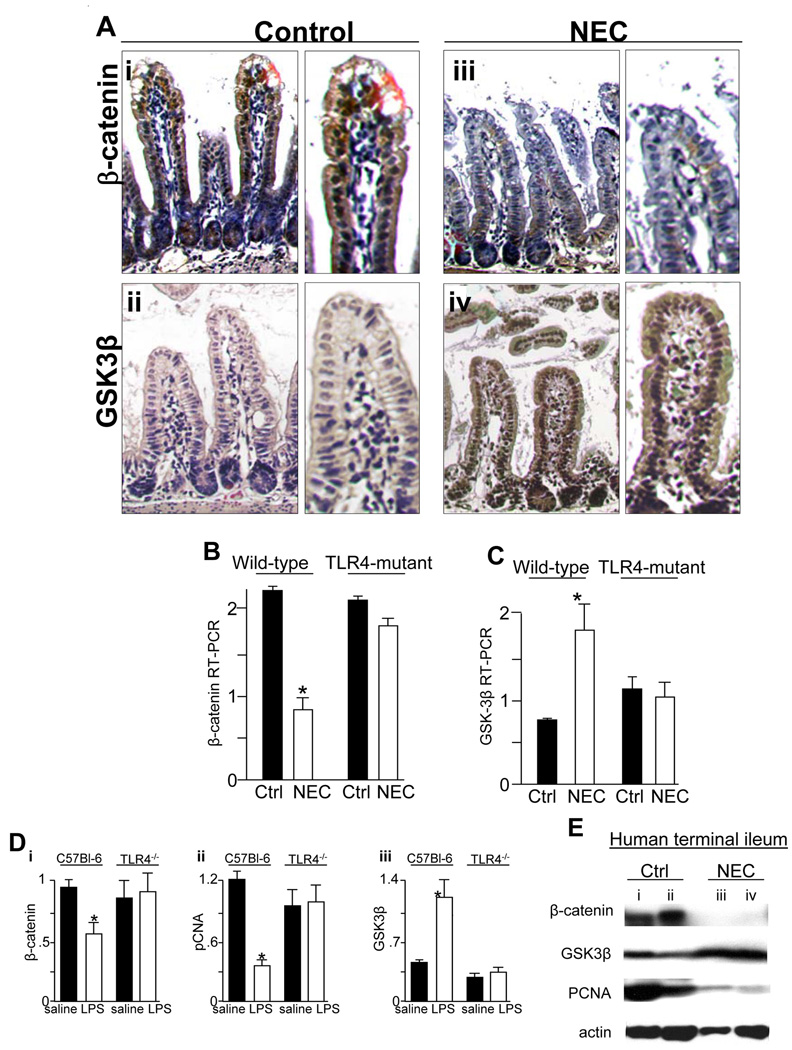
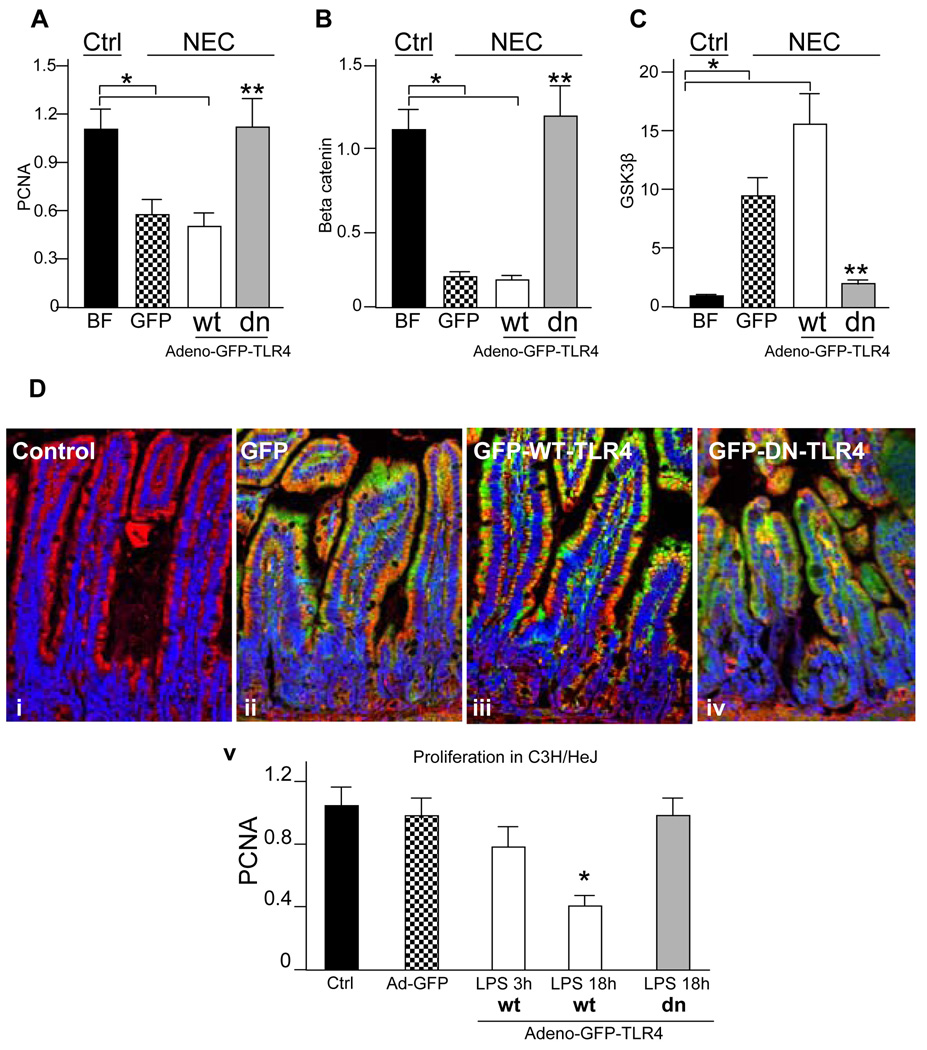
Methods: Enterocyte proliferation was detected in IEC-6 cells or in ileum or colon from wild-type, TLR4-mutant, or TLR4(-/-) mice after induction of NEC or endotoxemia. beta-Catenin signaling was assessed by cell fractionation or immunoconfocal microscopy to detect its nuclear translocation. Activation and inhibition of beta-catenin were achieved via cDNA or small interfering RNA, respectively. TLR4 in the intestinal mucosa was inhibited with adenoviruses expressing dominant-negative TLR4.
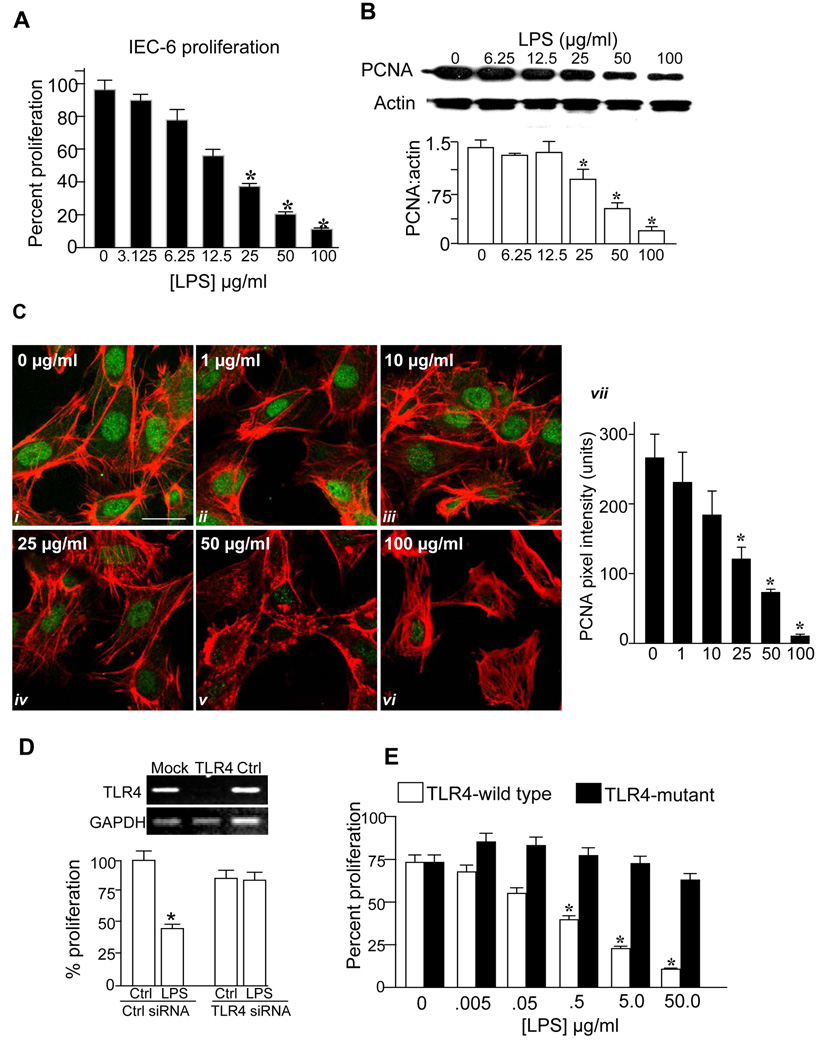
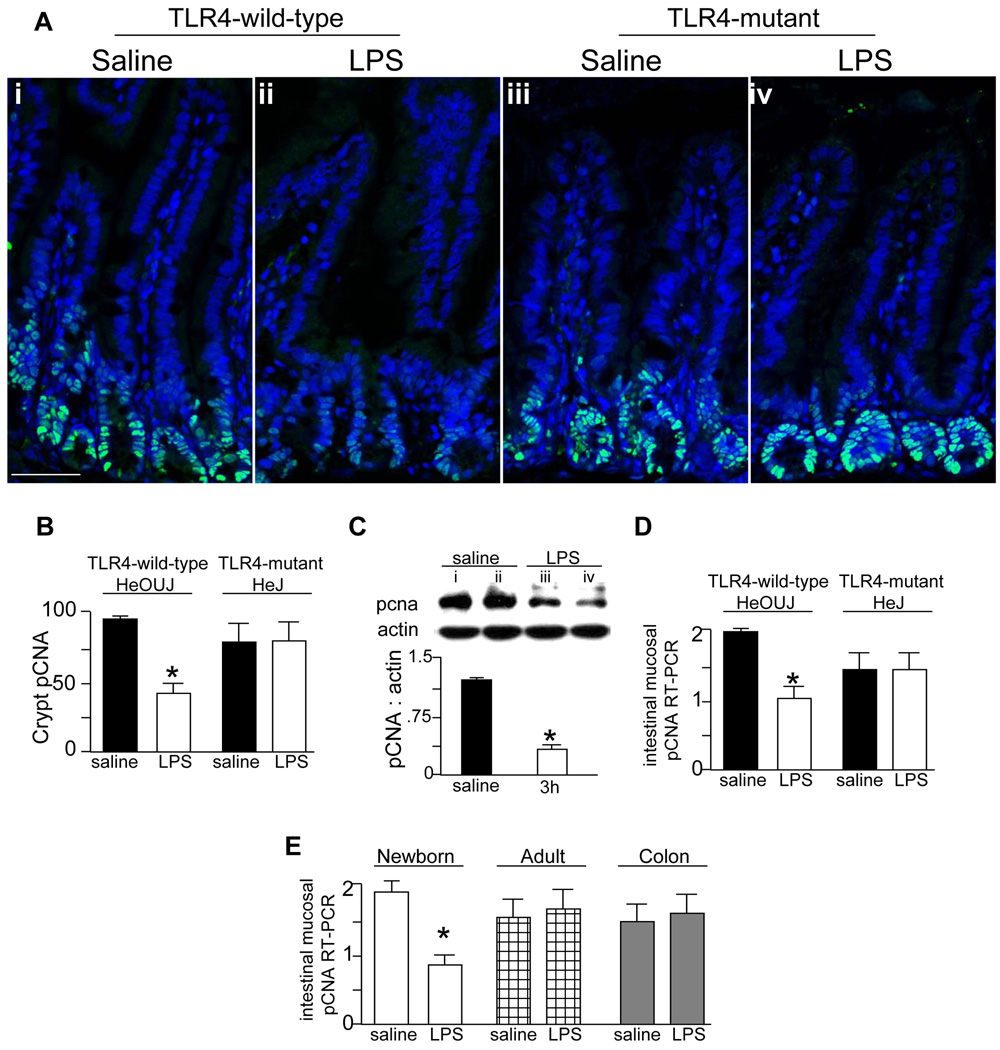
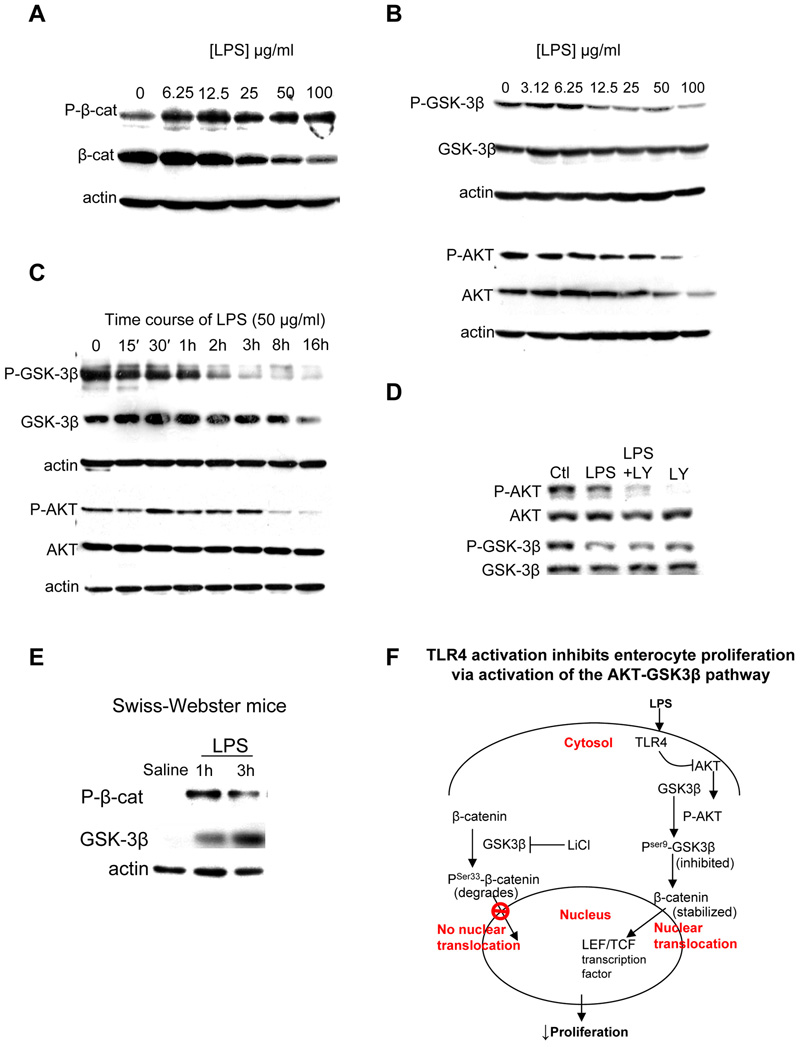
Results: TLR4 activation significantly impaired enterocyte proliferation in the ileum but not colon in newborn but not adult mice and in IEC-6 enterocytes. beta-Catenin activation reversed these effects in vitro. To determine the mechanisms involved, TLR4 activation phosphorylated the upstream inhibitory kinase GSK3beta, causing beta-catenin degradation. NEC in both mouse and humans was associated with decreased beta-catenin and increased mucosal GSK3beta expression. Strikingly, the inhibition of enterocyte beta-catenin signaling in NEC could be reversed, and enterocyte proliferation restored, through adenoviral-mediated inhibition of TLR4 signaling in the small intestinal mucosa.
Conclusion: We now report a novel pathway linking TLR4 with inhibition of beta-catenin signaling via GSK3beta activation, leading to reduced enterocyte proliferation in vitro and in vivo. These data provide additional insights into the pathogenesis of diseases of intestinal inflammation such as NEC.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest:
Xia-hua Shi – no conflicts exist; Chhinder P. Sodhi - – no conflicts exist ; Ward M. Richardson - – no conflicts exist; Steven C. Gribar - – no conflicts exist; Thomas Prindle Jr - – no conflicts exist; Maria Branca – no conflicts exist; Anthony Russo - – no conflicts exist; Congrong Ma - – no conflicts exist ; Richard Shapiro – no conflicts exist; Zachary Grant – no conflicts exist; David J. Hackam – no conflicts exist.
Transcript profiling – Not applicable
Writing assistance – Not applicable.
Figures
Comment in
-
The Ying and Yang of bacterial signaling in necrotizing enterocolitis.Gastroenterology. 2010 Jan;138(1):39-43. doi: 10.1053/j.gastro.2009.11.031. Epub 2009 Nov 21. Gastroenterology. 2010. PMID: 19932667 No abstract available.
References
-
- Gribar SCAR, Sodhi CP, Hackam DJ. The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J Leukoc Biol. 2008;83:493–498. - PubMed
-
- Anand RJ, Leaphart CL, Mollen KP, Hackam DJ. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27:124–133. - PubMed
-
- Grave GD, Nelson SA, Walker WA, Moss RL, Dvorak B, Hamilton FA, Higgins R, Raju TN. New therapies and preventive approaches for necrotizing enterocolitis: report of a research planning workshop. Pediatr Res. 2007;62:510–514. - PubMed
-
- Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem. 2004;279:24592–24600. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
