

Mitochondrial dysfunction increases allergic airway inflammation
- PMID: 19786549
- PMCID: PMC3028535
- DOI: 10.4049/jimmunol.0900228
Mitochondrial dysfunction increases allergic airway inflammation
Abstract
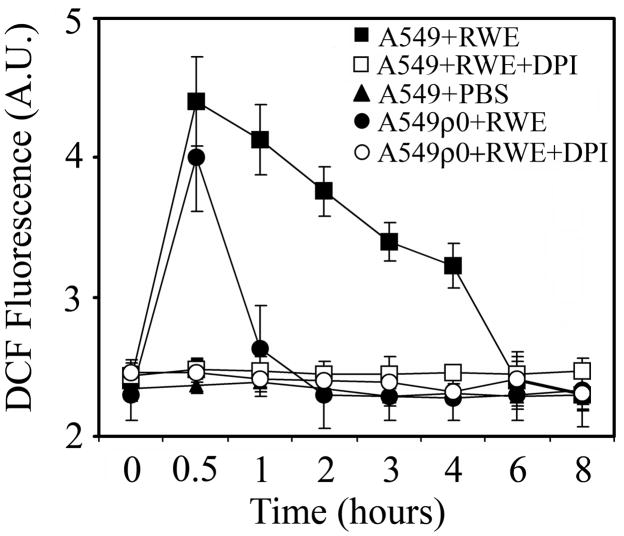
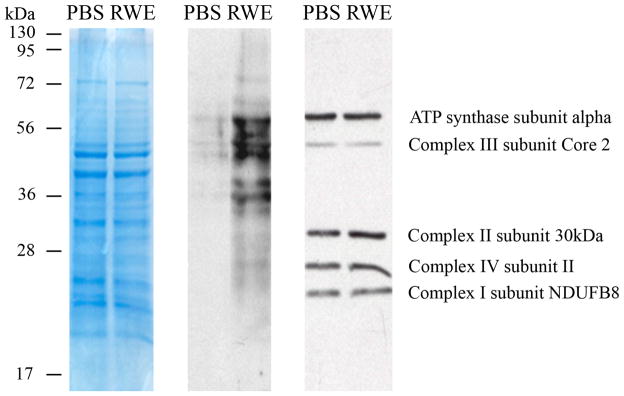
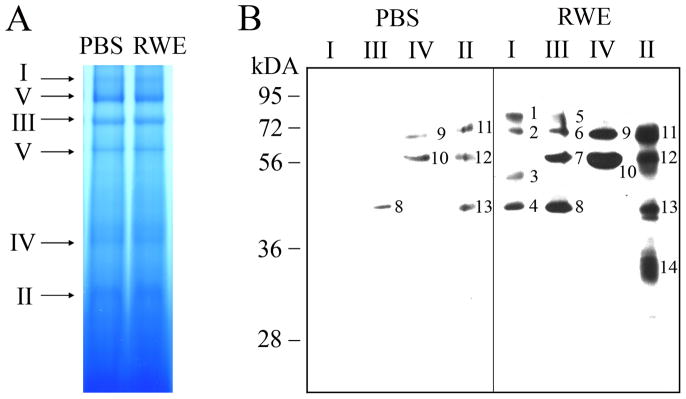
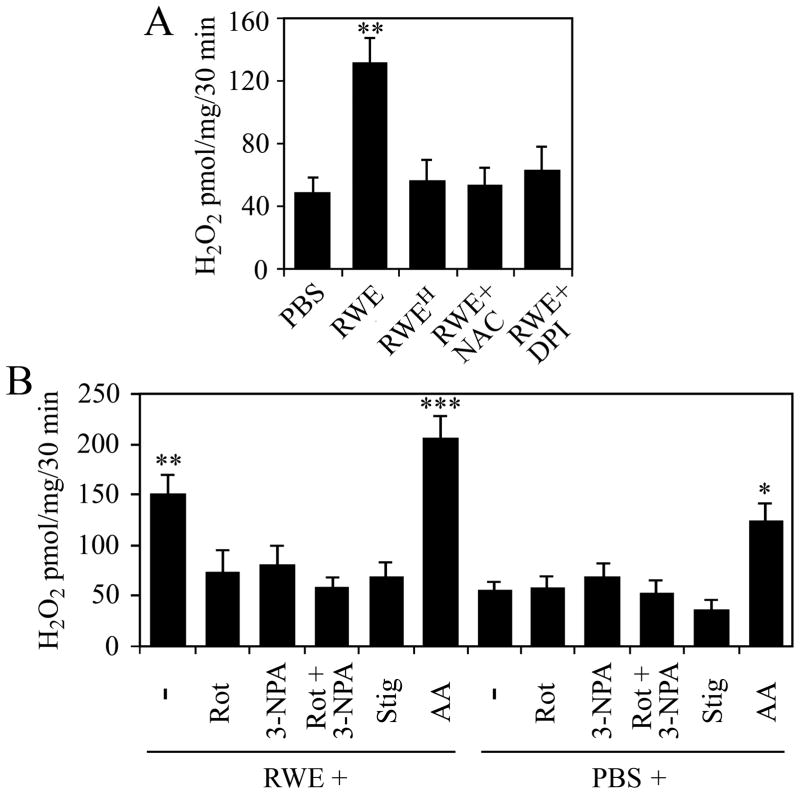
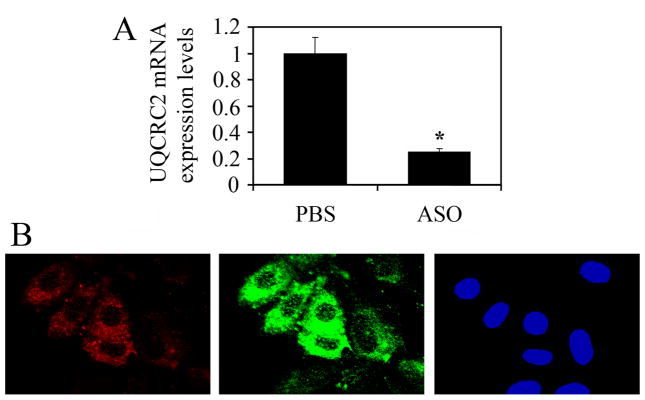
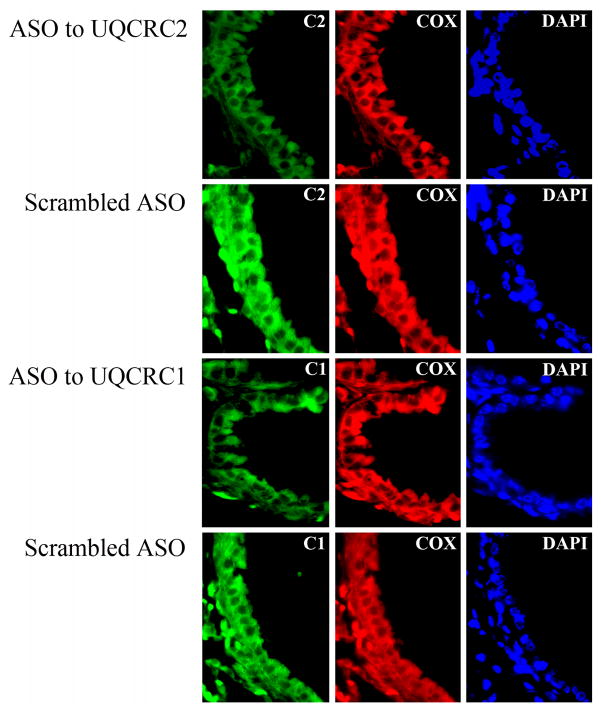
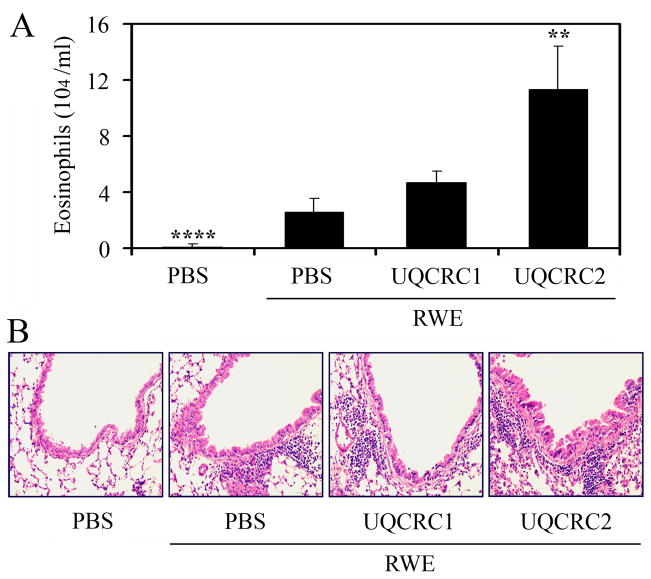
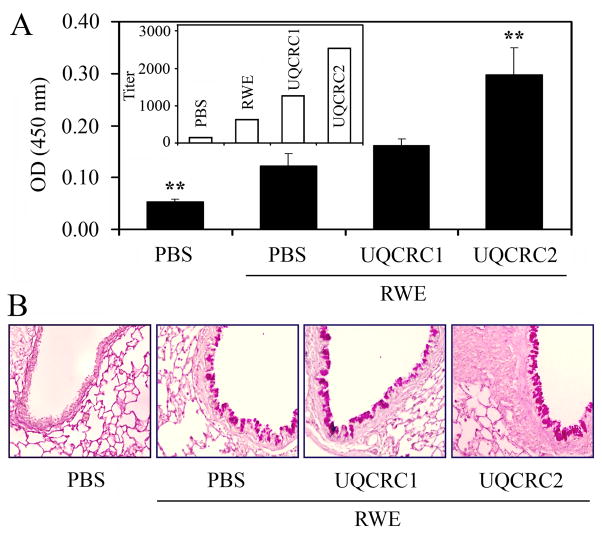
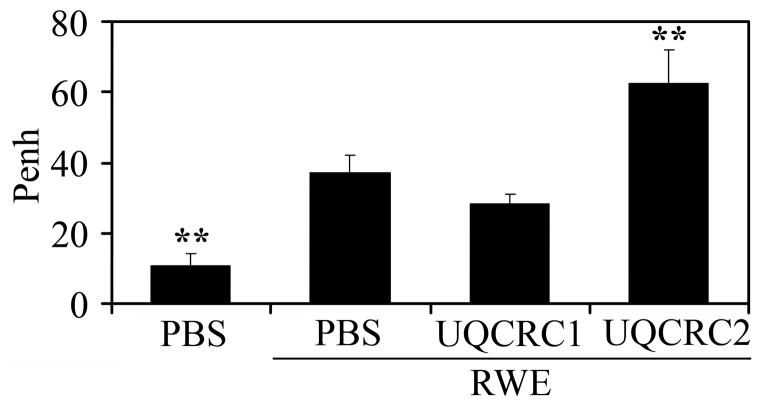
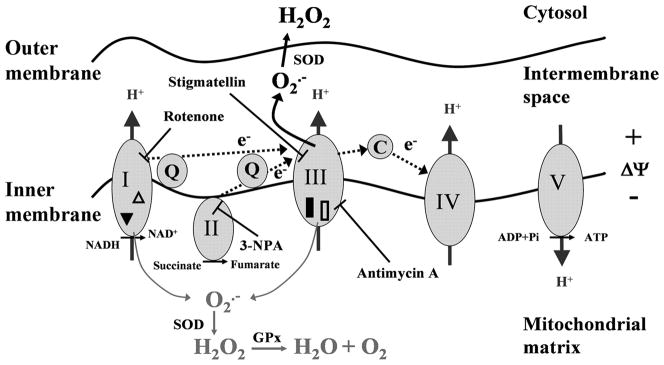
The prevalence of allergies and asthma among the world's population has been steadily increasing due to environmental factors. It has been described that exposure to ozone, diesel exhaust particles, or tobacco smoke exacerbates allergic inflammation in the lungs. These environmental oxidants increase the levels of cellular reactive oxygen species (ROS) and induce mitochondrial dysfunction in the airway epithelium. In this study, we investigated the involvement of preexisting mitochondrial dysfunction in the exacerbation of allergic airway inflammation. After cellular oxidative insult induced by ragweed pollen extract (RWE) exposure, we have identified nine oxidatively damaged mitochondrial respiratory chain-complex and associated proteins. Out of these, the ubiquinol-cytochrome c reductase core II protein (UQCRC2) was found to be implicated in mitochondrial ROS generation from respiratory complex III. Mitochondrial dysfunction induced by deficiency of UQCRC2 in airway epithelium of sensitized BALB/c mice prior the RWE challenge increased the Ag-induced accumulation of eosinophils, mucin levels in the airways, and bronchial hyperresponsiveness. Deficiency of UQCRC1, another oxidative damage-sensitive complex III protein, did not significantly alter cellular ROS levels or the intensity of RWE-induced airway inflammation. These observations suggest that preexisting mitochondrial dysfunction induced by oxidant environmental pollutants is responsible for the severe symptoms in allergic airway inflammation. These data also imply that mitochondrial defects could be risk factors and may be responsible for severe allergic disorders in atopic individuals.
Figures
References
-
- Bowler RP. Oxidative stress in the pathogenesis of asthma. Curr Allergy Asthma Rep. 2004;4:116–122. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
