

De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies
- PMID: 19786696
- PMCID: PMC2754324
- DOI: 10.1212/WNL.0b013e3181b9cebc
De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies
Abstract
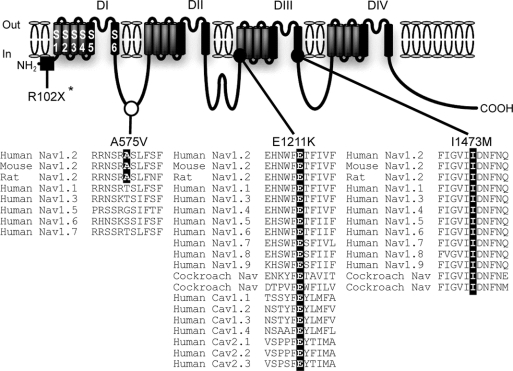
Background: Mutations of voltage-gated sodium channel alpha(II) gene, SCN2A, have been described in a wide spectrum of epilepsies. While inherited SCN2A mutations have been identified in multiple mild epilepsy cases, a de novo SCN2A-R102X mutation, which we previously reported in a patient with sporadic intractable childhood localization-related epilepsy, remains unique. To validate the involvement of de novo SCN2A mutations in the etiology of intractable epilepsies, we sought to identify additional instances.
Methods: We performed mutational analyses on SCN2A in 116 patients with severe myoclonic epilepsy in infancy, infantile spasms, and other types of intractable childhood partial and generalized epilepsies and did whole-cell patch-clamp recordings on Na(v)1.2 channels containing identified mutations.
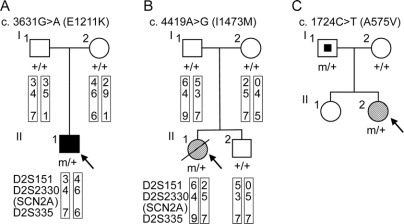
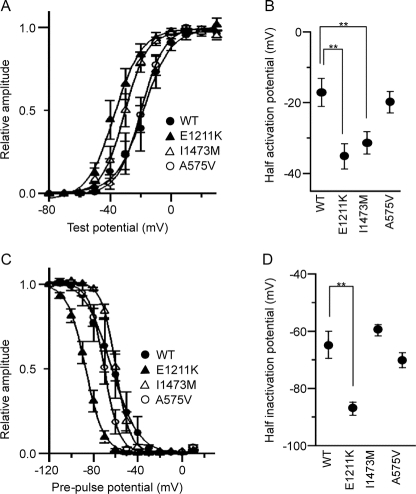
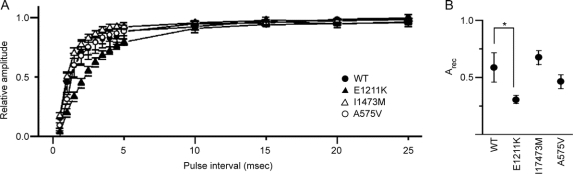
Results: We discovered 2 additional de novo SCN2A mutations. One mutation, SCN2A-E1211K, was identified in a patient with sporadic infantile spasms. SCN2A-E1211K produced channels with altered electrophysiologic properties compatible with both augmented (an approximately 18-mV hyperpolarizing shift in the voltage dependence of activation) and reduced (an approximately 22-mV hyperpolarizing shift in the voltage dependence of steady-state inactivation and a slowed recovery from inactivation) channel activities. The other de novo mutation, SCN2A-I1473M, was identified in a patient with sporadic neonatal epileptic encephalopathy. SCN2A-I1473M caused an approximately 14-mV hyperpolarizing shift in the voltage dependence of activation.
Conclusions: The identified de novo mutations SCN2A-E1211K, -I1473M, and -R102X indicate that SCN2A is an etiologic candidate underlying a variety of intractable childhood epilepsies. The phenotypic variations among patients might be due to the different electrophysiologic properties of mutant channels.
Figures
Comment in
-
De novo nonsense mutations in the sodium channel gene, SCN2A, in sporadic intractable epilepsy.Clin Genet. 2010 Jun;77(6):538-40. doi: 10.1111/j.1399-0004.2010.01396_3.x. Epub 2010 Mar 1. Clin Genet. 2010. PMID: 20236112 No abstract available.
References
-
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. - PubMed
-
- Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 2000;24:343–345. - PubMed
-
- Sugawara T, Mazaki-Miyazaki E, Ito M, et al. Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology 2001;57:703–705. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases