

Exploration of uncharted regions of the protein universe
- PMID: 19787035
- PMCID: PMC2744874
- DOI: 10.1371/journal.pbio.1000205
Exploration of uncharted regions of the protein universe
Abstract
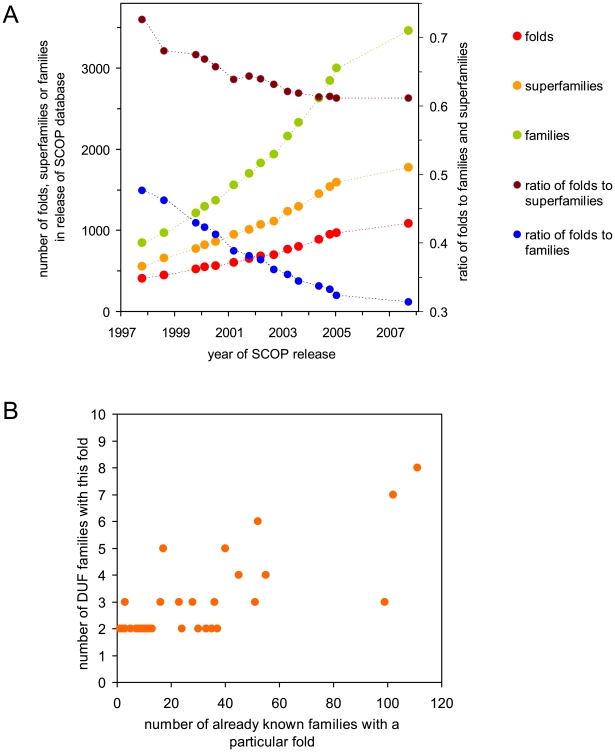
The genome projects have unearthed an enormous diversity of genes of unknown function that are still awaiting biological and biochemical characterization. These genes, as most others, can be grouped into families based on sequence similarity. The PFAM database currently contains over 2,200 such families, referred to as domains of unknown function (DUF). In a coordinated effort, the four large-scale centers of the NIH Protein Structure Initiative have determined the first three-dimensional structures for more than 250 of these DUF families. Analysis of the first 248 reveals that about two thirds of the DUF families likely represent very divergent branches of already known and well-characterized families, which allows hypotheses to be formulated about their biological function. The remainder can be formally categorized as new folds, although about one third of these show significant substructure similarity to previously characterized folds. These results infer that, despite the enormous increase in the number and the diversity of new genes being uncovered, the fold space of the proteins they encode is gradually becoming saturated. The previously unexplored sectors of the protein universe appear to be primarily shaped by extreme diversification of known protein families, which then enables organisms to evolve new functions and adapt to particular niches and habitats. Notwithstanding, these DUF families still constitute the richest source for discovery of the remaining protein folds and topologies.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





Comment in
-
Charting an unknown protein universe.PLoS Biol. 2009 Sep 29;7(9):e1000206. doi: 10.1371/journal.pbio.1000206. PLoS Biol. 2009. PMID: 20076754 Free PMC article. No abstract available.
References
-
- Gerdes S, Edwards R, Kubal M, Fonstein M, Stevens R, et al. Essential genes on metabolic maps. Curr Opin Biotechnol. 2006;17:448–456. - PubMed
-
- Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol. 2005;55:137–149. - PubMed
-
- Fan J. B, Chee M. S, Gunderson K. L. Highly parallel genomic assays. Nat Rev Genet. 2006;7:632–644. - PubMed
-
- Seng K. C, Seng C. K. The success of the genome-wide association approach: a brief story of a long struggle. Eur J Hum Genet. 2008;16:554–564. - PubMed
