

Using non-homogeneous models of nucleotide substitution to identify host shift events: application to the origin of the 1918 'Spanish' influenza pandemic virus
- PMID: 19787384
- PMCID: PMC2772961
- DOI: 10.1007/s00239-009-9282-x
Using non-homogeneous models of nucleotide substitution to identify host shift events: application to the origin of the 1918 'Spanish' influenza pandemic virus
Abstract
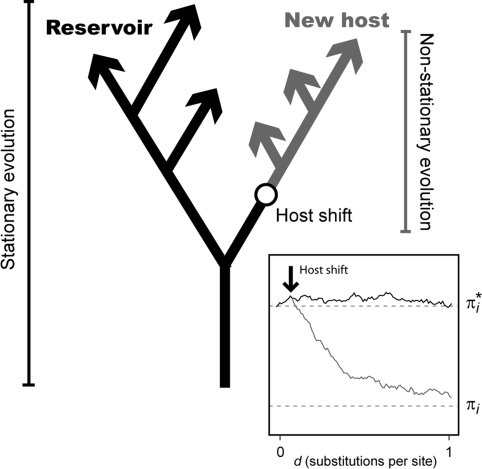
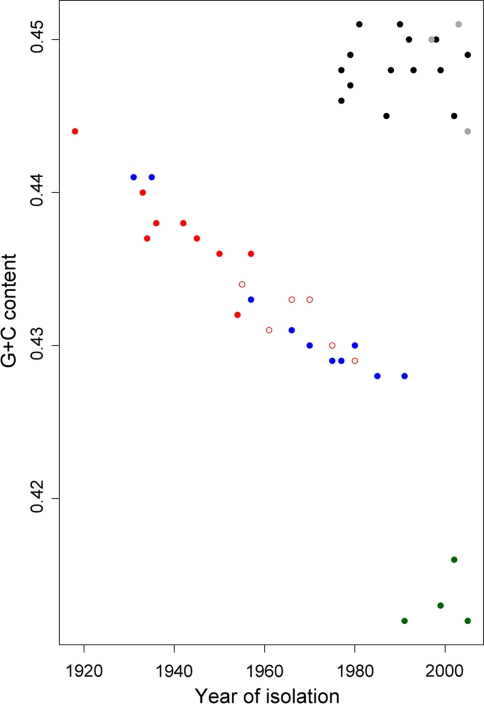
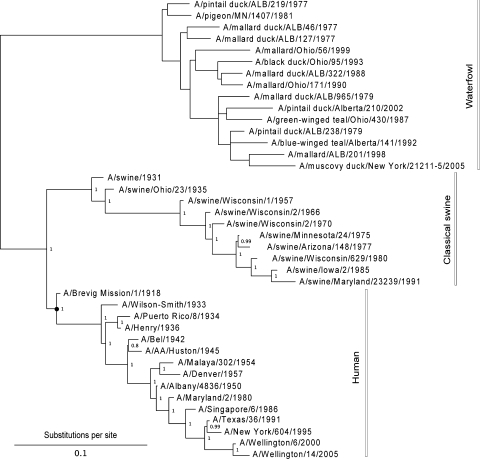
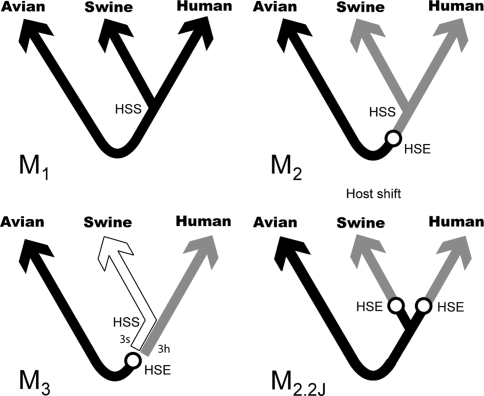
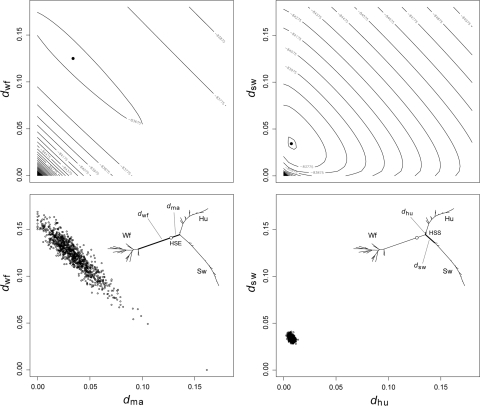
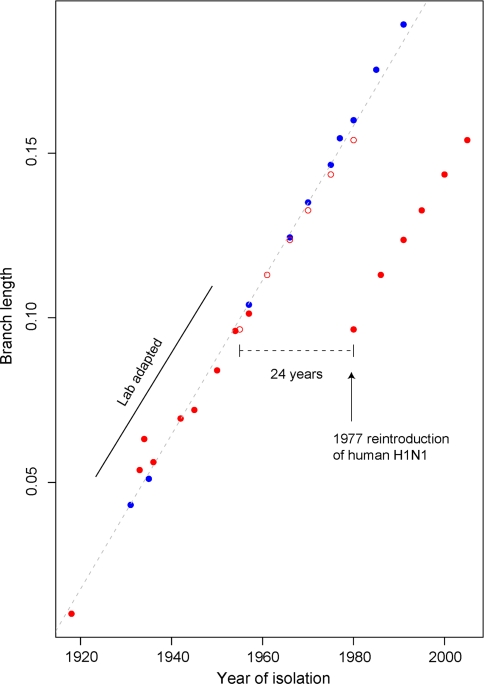
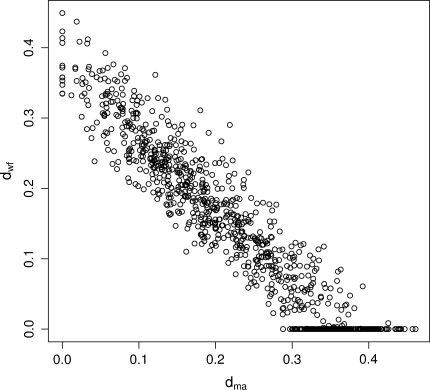
Nonhomogeneous Markov models of nucleotide substitution have received scant attention. Here we explore the possibility of using nonhomogeneous models to identify host shift nodes along phylogenetic trees of pathogens evolving in different hosts. It has been noticed that influenza viruses show marked differences in nucleotide composition in human and avian hosts. We take advantage of this fact to identify the host shift event that led to the 1918 'Spanish' influenza. This disease killed over 50 million people worldwide, ranking it as the deadliest pandemic in recorded history. Our model suggests that the eight RNA segments which eventually became the 1918 viral genome were introduced into a mammalian host around 1882-1913. The viruses later diverged into the classical swine and human H1N1 influenza lineages around 1913-1915. The last common ancestor of human strains dates from February 1917 to April 1918. Because pigs are more readily infected with avian influenza viruses than humans, it would seem that they were the original recipient of the virus. This would suggest that the virus was introduced into humans sometime between 1913 and 1918.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
