

Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype
- PMID: 19789634
- PMCID: PMC2747278
- DOI: 10.1371/journal.pone.0007232
Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype
Abstract
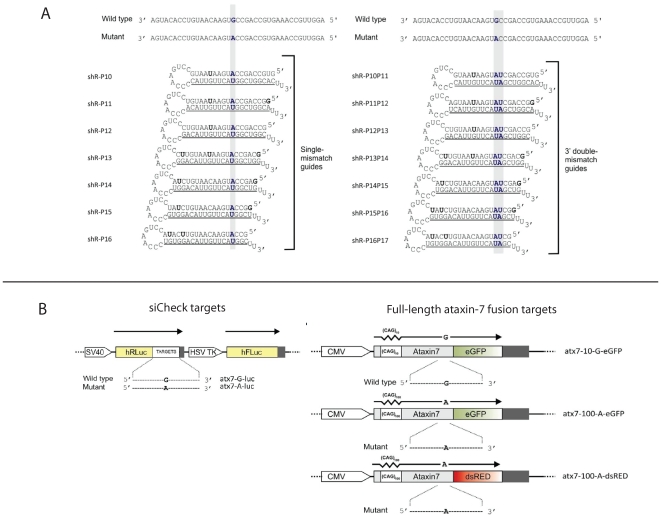
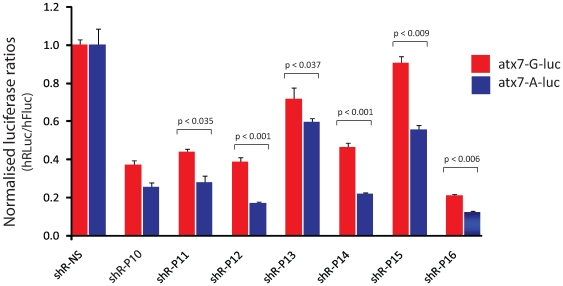
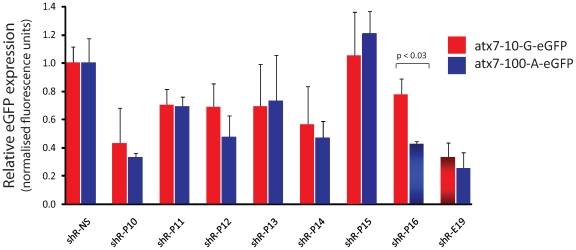
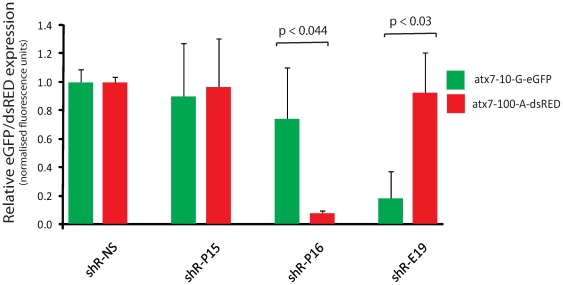
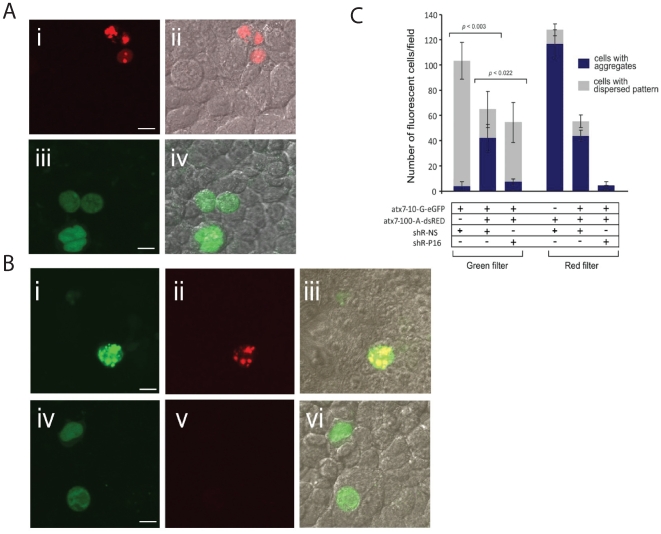
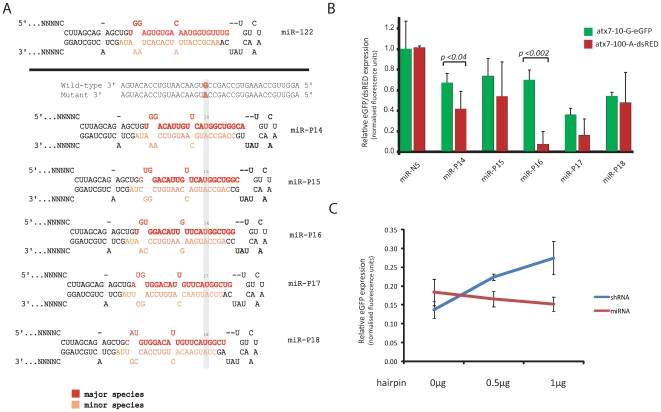
Spinocerebellar ataxia type 7 is a polyglutamine disorder caused by an expanded CAG repeat mutation that results in neurodegeneration. Since no treatment exists for this chronic disease, novel therapies such post-transcriptional RNA interference-based gene silencing are under investigation, in particular those that might enable constitutive and tissue-specific silencing, such as expressed hairpins. Given that this method of silencing can be abolished by the presence of nucleotide mismatches against the target RNA, we sought to identify expressed RNA hairpins selective for silencing the mutant ataxin-7 transcript using a linked SNP. By targeting both short and full-length tagged ataxin-7 sequences, we show that mutation-specific selectivity can be obtained with single nucleotide mismatches to the wild-type RNA target incorporated 3' to the centre of the active strand of short hairpin RNAs. The activity of the most effective short hairpin RNA incorporating the nucleotide mismatch at position 16 was further studied in a heterozygous ataxin-7 disease model, demonstrating significantly reduced levels of toxic mutant ataxin-7 protein with decreased mutant protein aggregation and retention of normal wild-type protein in a non-aggregated diffuse cellular distribution. Allele-specific mutant ataxin7 silencing was also obtained with the use of primary microRNA mimics, the most highly effective construct also harbouring the single nucleotide mismatch at position 16, corroborating our earlier findings. Our data provide understanding of RNA interference guide strand anatomy optimised for the allele-specific silencing of a polyglutamine mutation linked SNP and give a basis for the use of allele-specific RNA interference as a viable therapeutic approach for spinocerebellar ataxia 7.
Conflict of interest statement
Figures
References
-
- Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, et al. On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol. 1994;88:277–286. - PubMed
-
- David G, Abbas N, Stevanin G, Dürr A, Yvert G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. - DOI - PubMed
-
- Bryer A, Krause A, Bill P, Davids V, Bryant D, et al. The hereditary adult-onset ataxias in South Africa. J Neurol Sci. 2003;216:47–54. - PubMed
-
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. - DOI - PubMed
-
- Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci. 2007;27:2483–2492. doi: 10.1523/JNEUROSCI.5453-06.2007. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
