

Nrf2: friend or foe for chemoprevention?
- PMID: 19793802
- PMCID: PMC2802668
- DOI: 10.1093/carcin/bgp231
Nrf2: friend or foe for chemoprevention?
Abstract
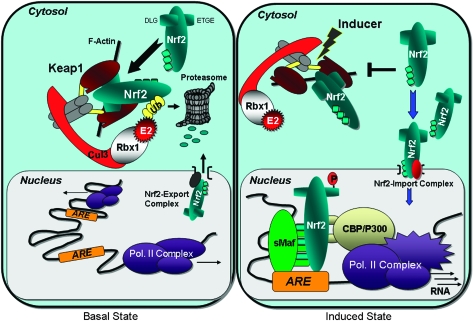
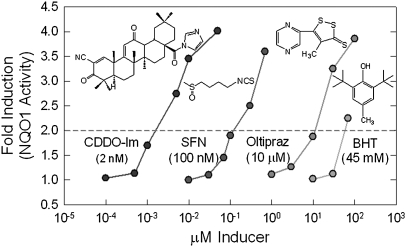
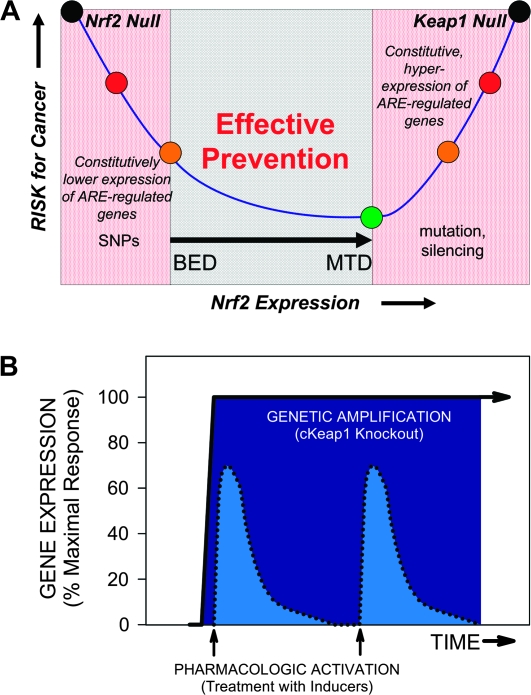
Health reflects the ability of an organism to adapt to stress. Stresses--metabolic, proteotoxic, mitotic, oxidative and DNA-damage stresses--not only contribute to the etiology of cancer and other chronic degenerative diseases but are also hallmarks of the cancer phenotype. Activation of the Kelch-like ECH-associated protein 1 (KEAP1)-NF-E2-related factor 2 (NRF2)-signaling pathway is an adaptive response to environmental and endogenous stresses and serves to render animals resistant to chemical carcinogenesis and other forms of toxicity, whilst disruption of the pathway exacerbates these outcomes. This pathway can be induced by thiol-reactive small molecules that demonstrate protective efficacy in preclinical chemoprevention models and in clinical trials. However, mutations and epigenetic modifications affecting the regulation and fate of NRF2 can lead to constitutive dominant hyperactivation of signaling that preserves rather than attenuates cancer phenotypes by providing selective resistance to stresses. This review provides a synopsis of KEAP1-NRF2 signaling, compares the impact of genetic versus pharmacologic activation and considers both the attributes and concerns of targeting the pathway in chemoprevention.
Figures
References
-
- Wattenberg LW. Inhibition of carcinogenic and toxic effects of polycyclic aromatic hydrocarbons by phenolic antioxidants and ethoxyquin. J. Natl Cancer Inst. 1972;48:1425–1430. - PubMed
-
- Benson AM, et al. Elevation of hepatic glutathione S-transferase activities and protection against mutagenic metabolites of benzo(a)pyrene by dietary antioxidants. Cancer Res. 1978;38:4486–4495. - PubMed
-
- Benson AM, et al. Elevation of extrahepatic glutathione S-transferase and epoxide hydratase activities by 2(3)-tert-butyl-4-hydroxyanisole. Cancer Res. 1979;39:2971–2977. - PubMed
-
- Talalay P, et al. Molecular mechanisms in protection against carcinogenesis. In: Cory JG, Szventivani A, editors. Cancer Biology and Therapeutics. New York, NY: Plenum Press; 1987. pp. 197–216.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
