

Lipid oversupply, selective insulin resistance, and lipotoxicity: molecular mechanisms
- PMID: 19796706
- PMCID: PMC2868077
- DOI: 10.1016/j.bbalip.2009.09.015
Lipid oversupply, selective insulin resistance, and lipotoxicity: molecular mechanisms
Abstract
The accumulation of fat in tissues not suited for lipid storage has deleterious consequences on organ function, leading to cellular damage that underlies diabetes, heart disease, and hypertension. To combat these lipotoxic events, several therapeutics improve insulin sensitivity and/or ameliorate features of metabolic disease by limiting the inappropriate deposition of fat in peripheral tissues (i.e. thiazolidinediones, metformin, and statins). Recent advances in genomics and lipidomics have accelerated progress towards understanding the pathogenic events associated with the excessive production, underutilization, or inefficient storage of fat. Herein we review studies applying pharmacological or genetic strategies to manipulate the expression or activity of enzymes controlling lipid deposition, in order to gain a clearer understanding of the molecular mechanisms by which fatty acids contribute to metabolic disease.
Copyright (c) 2009. Published by Elsevier B.V.
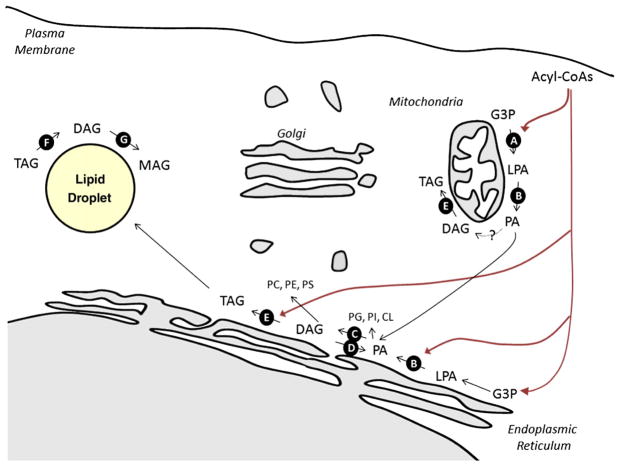
Figures



References
-
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. - PubMed
-
- CDC. US Center for Disease Control—National Diabetes Fact Sheet. Vol. 2009. 2009.
-
- CDC. US Center for Disease Control—Obesity Trends. Vol. 2009. 2009.
-
- Low S, Chin MC, Deurenberg-Yap M. Review on epidemic of obesity. Ann Acad Med Singapore. 2009;38:57–59. - PubMed
-
- WHO. World Health Organization - Diabetes Programme. Vol. 2009. 2009.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
