

Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia
- PMID: 19796717
- PMCID: PMC2818230
- DOI: 10.1016/j.bone.2009.09.016
Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia
Abstract
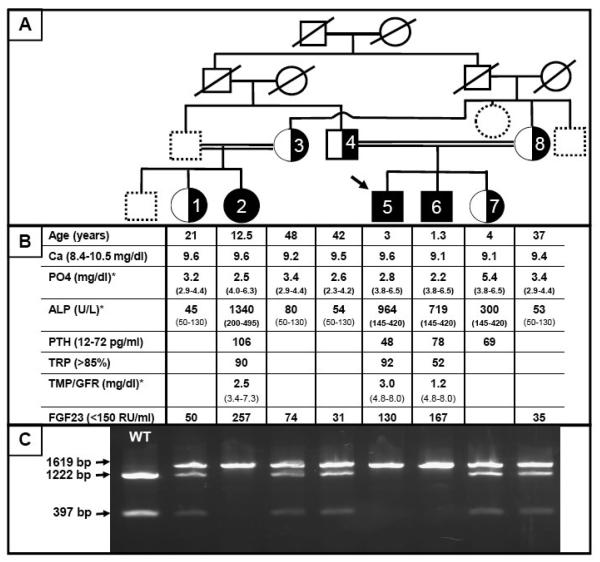
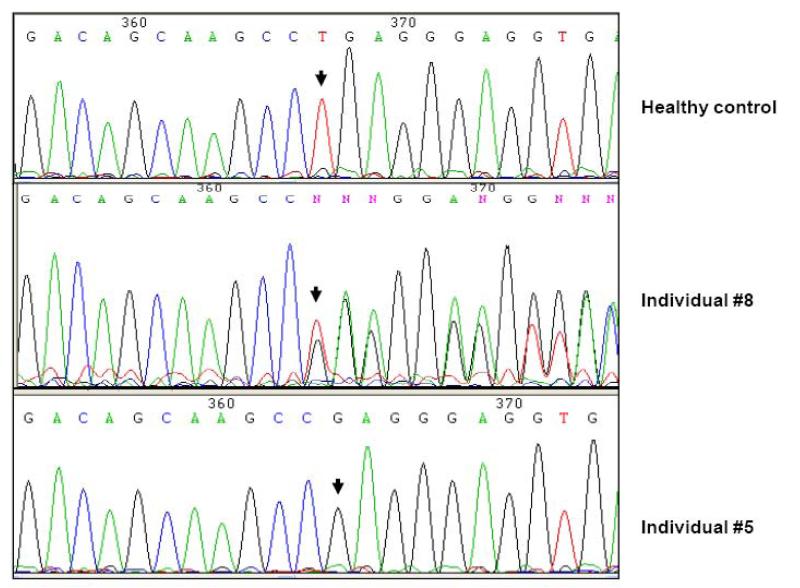
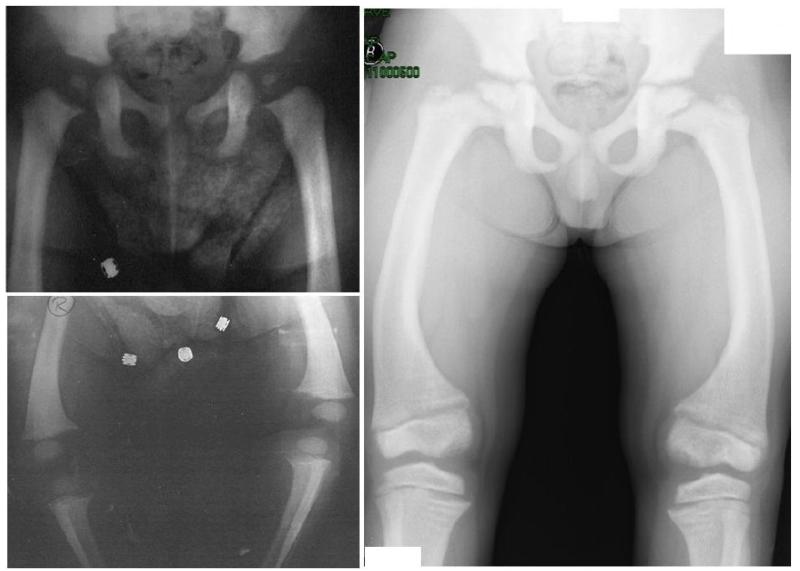
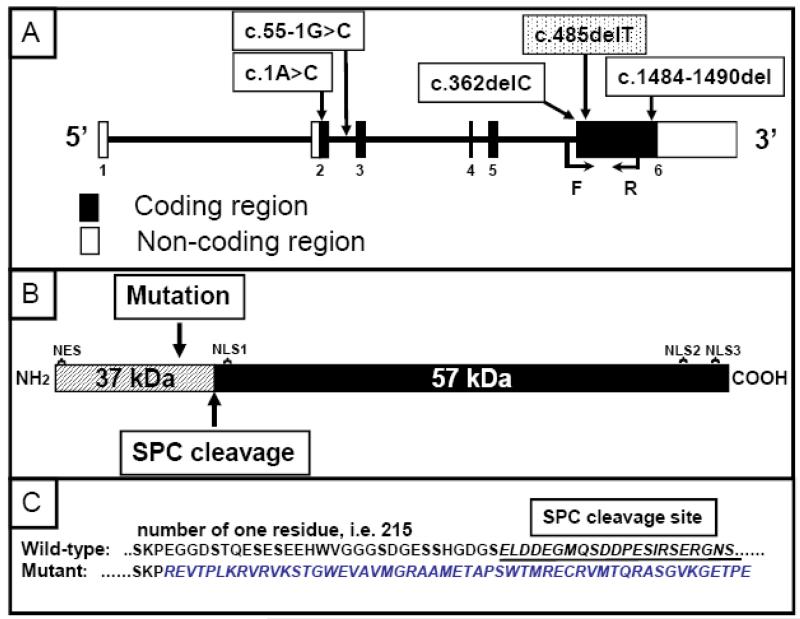
An autosomal recessive form of hypophosphatemia (ARHP) was recently shown to be caused by homozygous mutations in DMP1, the gene encoding dentin matrix protein-1 (DMP-1), a non-collagenous bone matrix protein with an important role in the development and mineralization of bone and teeth. Here, we describe a previously not reported consanguineous ARHP kindred in which the three affected individuals carry a novel homozygous DMP-1 mutation. The index case presented at the age of 3 years with bowing of his legs and showed hypophosphatemia due to insufficient renal phosphate retention. Serum alkaline phosphatase activity was elevated, with initially normal PTH. FGF23 was inappropriately normal at an older age while being treated with oral phosphate and 1,25(OH)(2)D. Similar clinical and biochemical findings, except for elevated FGF23 levels, were present in his 16-month-old brother and his 12.5-year-old female cousin; the parents of the three affected children are first-degree cousins. Nucleotide sequence analysis was performed on PCR-amplified exons encoding DMP-1 and flanking intronic regions. A novel homozygous frame-shift mutation (c.485Tdel; p.Glu163ArgfsX53) in exon 6 resulting in a premature stop codon was identified in all effected individuals. The parents and available unaffected siblings were heterozygous for c.485Tdel. Tooth growth and shape were normal for the index case, his affected brother and cousin, but their permanent and deciduous teeth displayed enlarged pulp chambers. The identified genetic mutation underscores the importance of DMP-1 mutations in the pathogenesis of ARHP. Furthermore, DMP-1 mutations appear to contribute, through yet unknown mechanisms, to tooth development.
(c) 2009 Elsevier Inc. All rights reserved.
Figures



Similar articles
-
A novel nonsense mutation in the DMP1 gene in a Japanese family with autosomal recessive hypophosphatemic rickets.J Bone Miner Metab. 2010 Sep;28(5):585-90. doi: 10.1007/s00774-010-0169-0. Epub 2010 Mar 9. J Bone Miner Metab. 2010. PMID: 20213538
-
Long-term clinical outcome and carrier phenotype in autosomal recessive hypophosphatemia caused by a novel DMP1 mutation.J Bone Miner Res. 2010 Oct;25(10):2165-74. doi: 10.1002/jbmr.105. J Bone Miner Res. 2010. PMID: 20499351 Free PMC article.
-
DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis.Nat Genet. 2006 Nov;38(11):1248-50. doi: 10.1038/ng1868. Epub 2006 Oct 8. Nat Genet. 2006. PMID: 17033625 Free PMC article.
-
Hypophosphatemic osteosclerosis, hyperostosis, and enthesopathy associated with novel homozygous mutations of DMP1 encoding dentin matrix protein 1 and SPP1 encoding osteopontin: The first digenic SIBLING protein osteopathy?Bone. 2020 Mar;132:115190. doi: 10.1016/j.bone.2019.115190. Epub 2019 Dec 13. Bone. 2020. PMID: 31843680 Free PMC article. Review.
-
Hereditary hypophosphatemias: new genes in the bone-kidney axis.Nephrology (Carlton). 2007 Aug;12(4):317-20. doi: 10.1111/j.1440-1797.2007.00824.x. Nephrology (Carlton). 2007. PMID: 17635744 Review.
Cited by
-
Enhancement of bone mineral density and body mass in newborn chickens by in ovo injection of ionic-hydroxyapatite nanoparticles of bacterial origin.J Mater Sci Mater Med. 2019 Jan 22;30(2):16. doi: 10.1007/s10856-018-6210-x. J Mater Sci Mater Med. 2019. PMID: 30671631
-
Genetics of Refractory Rickets: Identification of Novel PHEX Mutations in Indian Patients and a Literature Update.J Pediatr Genet. 2018 Jun;7(2):47-59. doi: 10.1055/s-0038-1624577. Epub 2018 Jan 28. J Pediatr Genet. 2018. PMID: 29707405 Free PMC article. Review.
-
Fibroblast growth factor 23: state of the field and future directions.Trends Endocrinol Metab. 2012 Dec;23(12):610-8. doi: 10.1016/j.tem.2012.07.002. Epub 2012 Aug 24. Trends Endocrinol Metab. 2012. PMID: 22921867 Free PMC article. Review.
-
New Therapies for Hypophosphatemia-Related to FGF23 Excess.Calcif Tissue Int. 2021 Jan;108(1):143-157. doi: 10.1007/s00223-020-00705-3. Epub 2020 Jun 5. Calcif Tissue Int. 2021. PMID: 32504139 Review.
-
Validation of a next-generation sequencing (NGS) panel to improve the diagnosis of X-linked hypophosphataemia (XLH) and other genetic disorders of renal phosphate wasting.Eur J Endocrinol. 2020 Nov;183(5):497-504. doi: 10.1530/EJE-20-0275. Eur J Endocrinol. 2020. PMID: 33107440 Free PMC article.
References
-
- ADHR Consortium, T. White KE, Evans WE, O'Riordan JLH, Speer MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meittinger T, Strom TM. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–8. - PubMed
-
- Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R, Giuca MR. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. 2006;7:61–6. - PubMed
-
- Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabedian M. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr. 2003;142:324–31. - PubMed
-
- Cohen S, Becker GL. Origin, diagnosis, and treatment of the dental manifestations of vitamin D-resistant rickets: review of the literature and report of case. J Am Dent Assoc. 1976;92:120–9. - PubMed
-
- Consortium TH. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
