

Dystroglycan matrix receptor function in cardiac myocytes is important for limiting activity-induced myocardial damage
- PMID: 19797173
- PMCID: PMC2783339
- DOI: 10.1161/CIRCRESAHA.109.199489
Dystroglycan matrix receptor function in cardiac myocytes is important for limiting activity-induced myocardial damage
Abstract
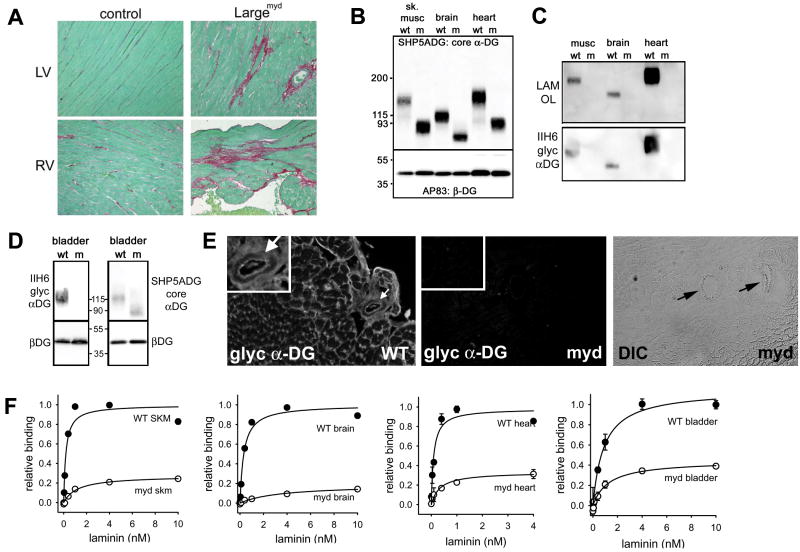
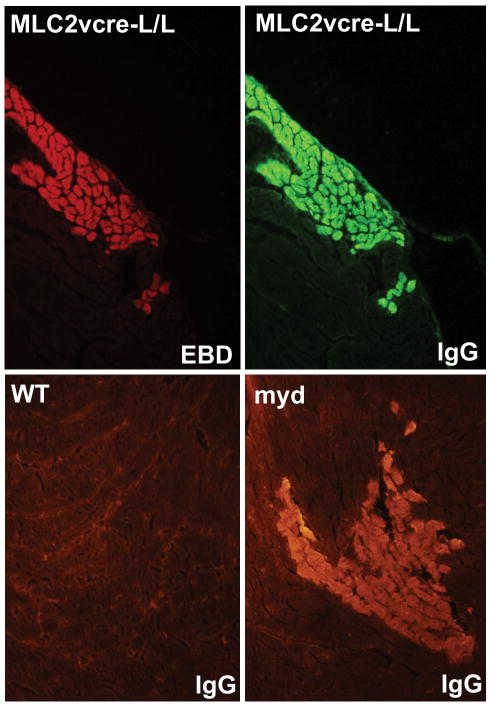
Rationale: Genetic mutations in a number of putative glycosyltransferases lead to the loss of glycosylation of dystroglycan and loss of its laminin-binding activity in genetic forms of human muscular dystrophy. Human patients and glycosylation defective myd mice develop cardiomyopathy with loss of dystroglycan matrix receptor function in both striated and smooth muscle.
Objective: To determine the functional role of dystroglycan in cardiac muscle and smooth muscle in the development of cardiomyopathy in muscular dystrophies.
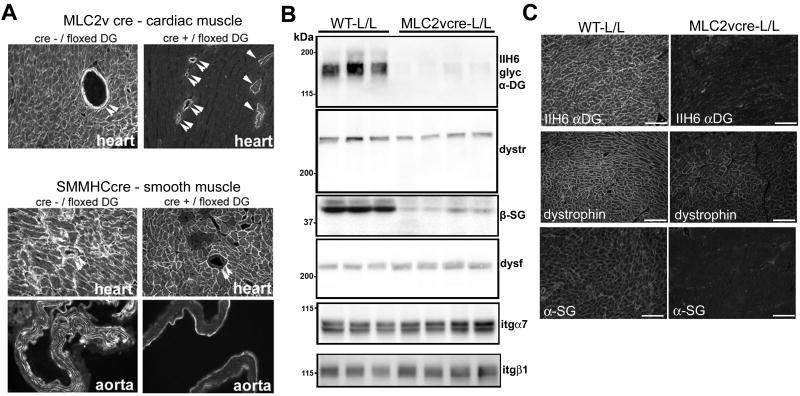
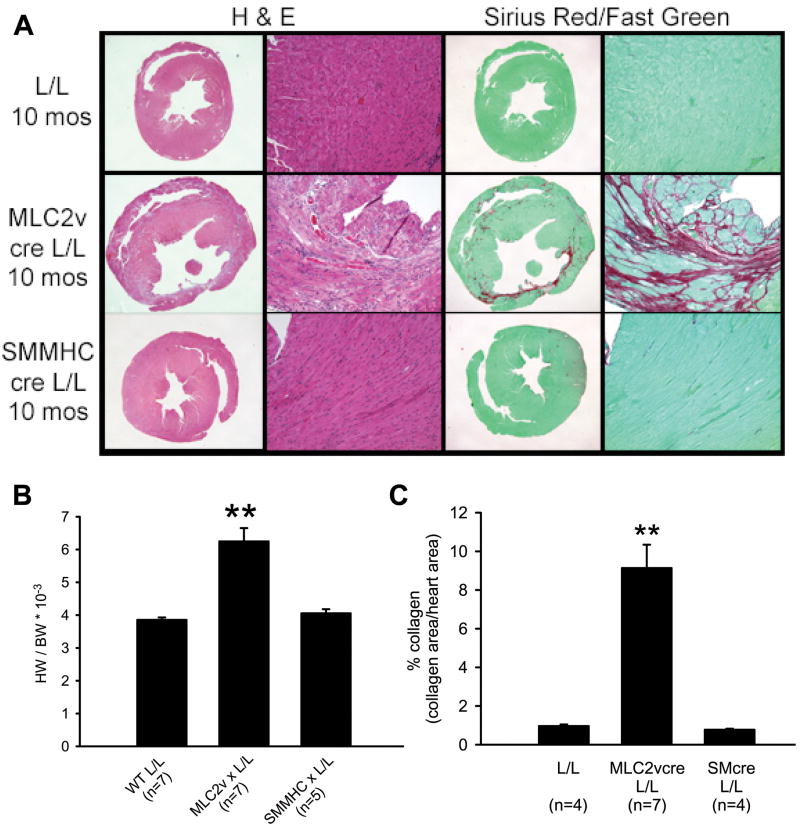
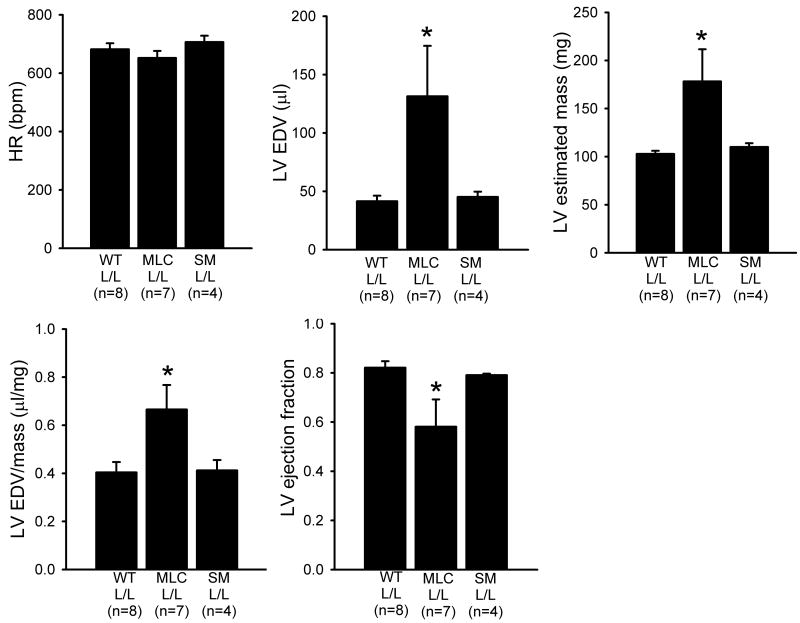
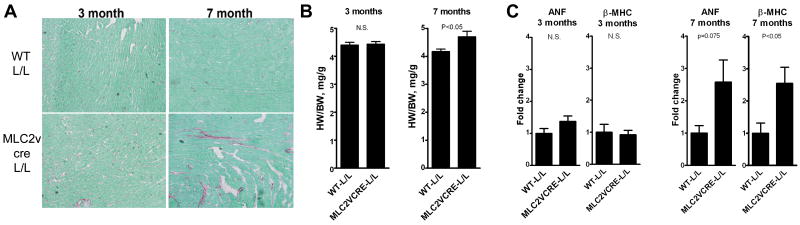
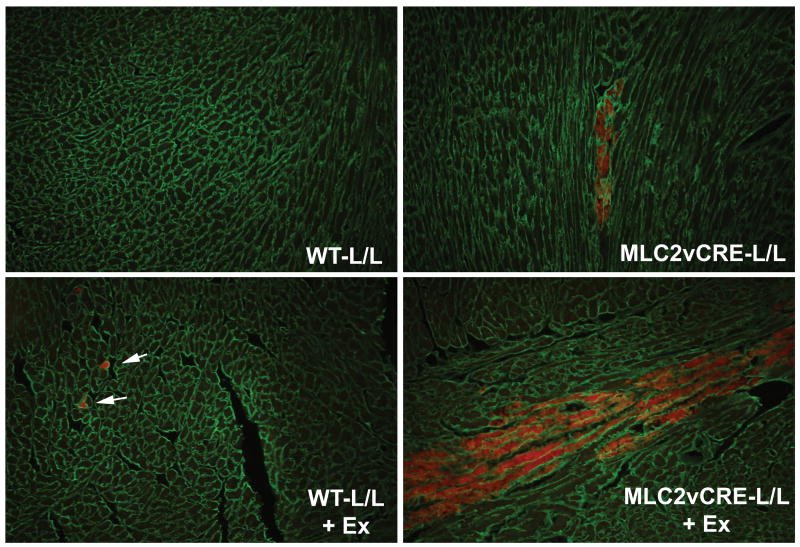
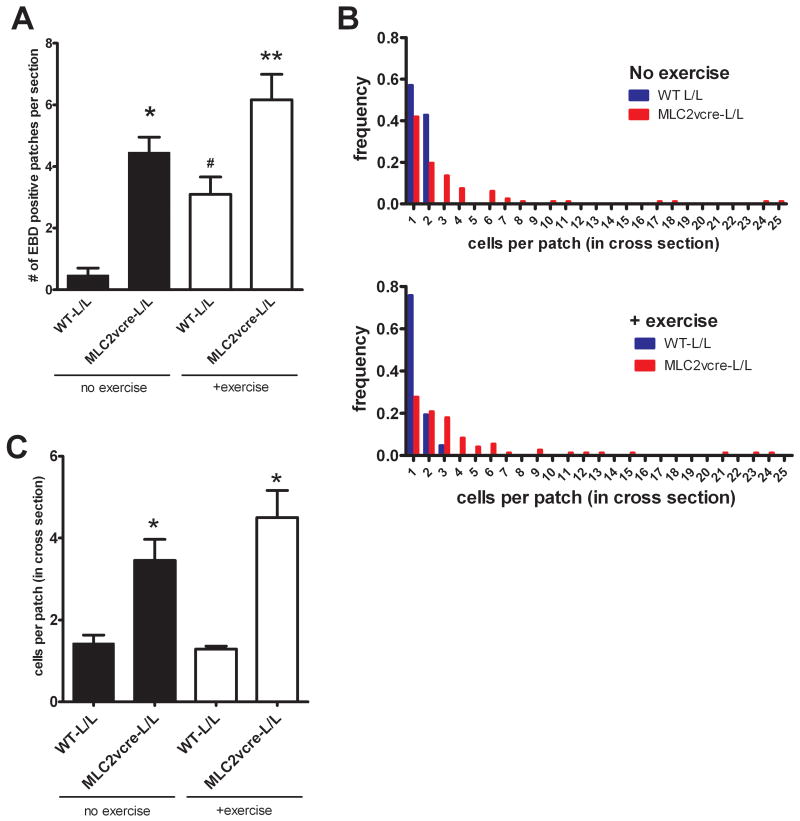
Methods and results: Using cre/lox-mediated gene targeting, we show here that loss of dystroglycan function in ventricular cardiac myocytes is sufficient to induce a progressive cardiomyopathy in mice characterized by focal cardiac fibrosis, increase in cardiac mass, and dilatation ultimately leading to heart failure. In contrast, disruption of dystroglycan in smooth muscle is not sufficient to induce cardiomyopathy. The specific loss of dystroglycan function in cardiac myocytes causes the accumulation of large, clustered patches of myocytes with membrane damage, which increase in number in response to exercise-induced cardiac stress, whereas exercised mice with normal dystroglycan expression accumulate membrane damage limited to individual myocytes.
Conclusions: Our findings suggest dystroglycan function as an extracellular matrix receptor in cardiac myocytes plays a primary role in limiting myocardial damage from spreading to neighboring cardiac myocytes, and loss of dystroglycan matrix receptor function in cardiac muscle cells is likely important in the development of cardiomyopathy in glycosylation-deficient muscular dystrophies.
Conflict of interest statement
Figures
References
-
- Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99(1):1–19. - PubMed
-
- Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98(4):465–474. - PubMed
-
- Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA, Campbell KP. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell. 2000;5(1):141–151. - PubMed
-
- Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, Politano L, Nigro G, Ventura C, Abbondanza C, Molinari AM, Acampora D, Nishimura M, Hayashizaki Y, Puca GA. Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet. 1997;6(4):601–607. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
