

Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction
- PMID: 19797174
- PMCID: PMC2783369
- DOI: 10.1161/CIRCRESAHA.109.199471
Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction
Abstract
Rationale: Interferon-gamma-inducible protein (IP)-10/CXCL10, an angiostatic and antifibrotic chemokine with an important role in T-cell trafficking, is markedly induced in myocardial infarcts, and may regulate the reparative response.
Objective: To study the role of IP-10 in cardiac repair and remodeling.
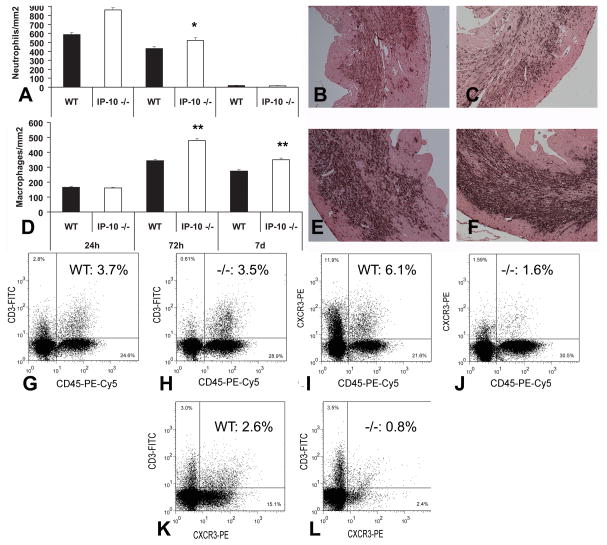
Methods and results: We studied cardiac repair in IP-10-null and wild-type (WT) mice undergoing reperfused infarction protocols and examined the effects of IP-10 on cardiac fibroblast function. IP-10-deficient and WT animals had comparable acute infarct size. However, the absence of IP-10 resulted in a hypercellular early reparative response and delayed contraction of the scar. Infarcted IP-10(-/-) hearts exhibited accentuated early dilation, followed by rapid wall thinning during infarct maturation associated with systolic dysfunction. Although IP-10-null and WT mice had comparable cytokine expression, the absence of IP-10 was associated with marked alterations in the cellular content of the infarct. IP-10(-/-) infarcts had more intense infiltration with CD45(+) leukocytes, Mac-2(+) macrophages, and alpha-smooth muscle actin (alpha-SMA)(+) myofibroblasts than WT infarcts but exhibited reduced recruitment of the subpopulations of leukocytes, T lymphocytes and alpha-SMA(+) cells that expressed CXCR3, the IP-10 receptor. IP-10 did not modulate cardiac fibroblast proliferation and apoptosis but significantly inhibited basic fibroblast growth factor-induced fibroblast migration. In addition, IP-10 enhanced growth factor-mediated wound contraction in fibroblast-populated collagen lattices.
Conclusions: Endogenous IP-10 is an essential inhibitory signal that regulates the cellular composition of the healing infarct and promotes wound contraction, attenuating adverse remodeling. IP-10-mediated actions may be due, at least in part, to direct effects on fibroblast migration and function.
Figures





References
-
- Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32. - PubMed
-
- Frangogiannis NG. Chemokines in ischemia and reperfusion. Thromb Haemost. 2007;97:738–747. - PubMed
-
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
