

Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes
- PMID: 19797425
- PMCID: PMC2802203
- DOI: 10.1093/cvr/cvp324
Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes
Abstract
Aims: Calmodulin (CaM) regulates Na+ channel gating through binding to an IQ-like motif in the C-terminus. Ca2+/CaM-dependent protein kinase II (CaMKII) regulates Ca2+ handling, and chronic overactivity of CaMKII is associated with left ventricular hypertrophy and dysfunction and lethal arrhythmias. However, the acute effects of Ca2+/CaM and CaMKII on cardiac Na+ channels are not fully understood.
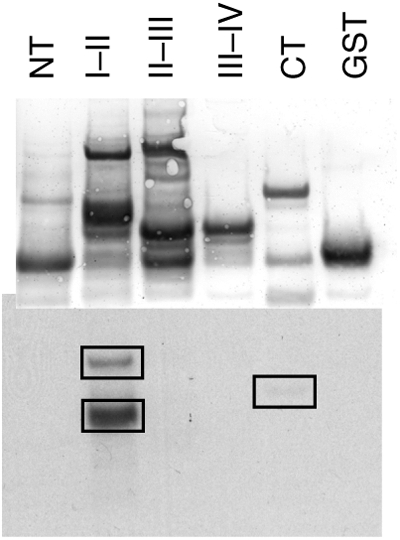
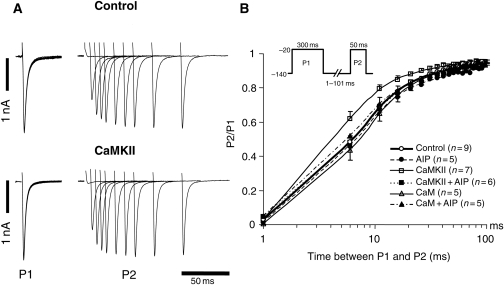
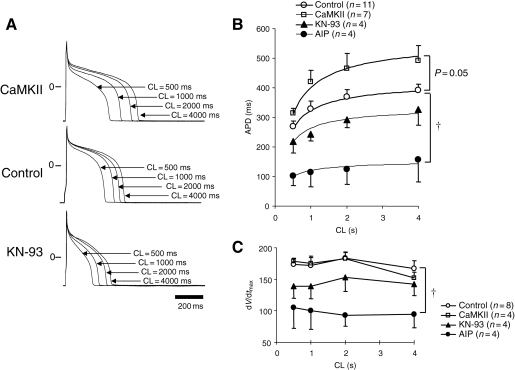
Methods and results: Purified Na(V)1.5-glutathione-S-transferase fusion peptides were phosphorylated in vitro by CaMKII predominantly on the I-II linker. Whole-cell voltage-clamp was used to measure Na+ current (I(Na)) in isolated guinea-pig ventricular myocytes in the absence or presence of CaM or CaMKII in the pipette solution. CaMKII shifted the voltage dependence of Na+ channel availability by approximately +5 mV, hastened recovery from inactivation, decreased entry into intermediate or slow inactivation, and increased persistent (late) current, but did not change I(Na) decay. These CaMKII-induced changes of Na+ channel gating were completely abolished by a specific CaMKII inhibitor, autocamtide-2-related inhibitory peptide (AIP). Ca2+/CaM alone reproduced the CaMKII-induced changes of I(Na) availability and the fraction of channels undergoing slow inactivation, but did not alter recovery from inactivation or the magnitude of the late current. Furthermore, the CaM-induced changes were also completely abolished by AIP. On the other hand, cAMP-dependent protein kinase A inhibitors did not abolish the CaM/CaMKII-induced alterations of I(Na) function.
Conclusion: Ca2+/CaM and CaMKII have distinct effects on the inactivation phenotype of cardiac Na+ channels. The differences are consistent with CaM-independent effects of CaMKII on cardiac Na+ channel gating.
Figures




Comment in
-
Calcium-dependent regulation of voltage-gated sodium channels in cardiac myocytes: just the beginning?Cardiovasc Res. 2010 Feb 1;85(3):411-2. doi: 10.1093/cvr/cvp398. Epub 2009 Dec 14. Cardiovasc Res. 2010. PMID: 20008474 No abstract available.
References
-
- Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. - PubMed
-
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. - PubMed
-
- Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–21. - PubMed
-
- Tan HL, Bink-Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
