

Paradoxical stimulation of cyclooxygenase-2 expression by glucocorticoids via a cyclic AMP response element in human amnion fibroblasts
- PMID: 19797430
- PMCID: PMC5419155
- DOI: 10.1210/me.2009-0201
Paradoxical stimulation of cyclooxygenase-2 expression by glucocorticoids via a cyclic AMP response element in human amnion fibroblasts
Abstract
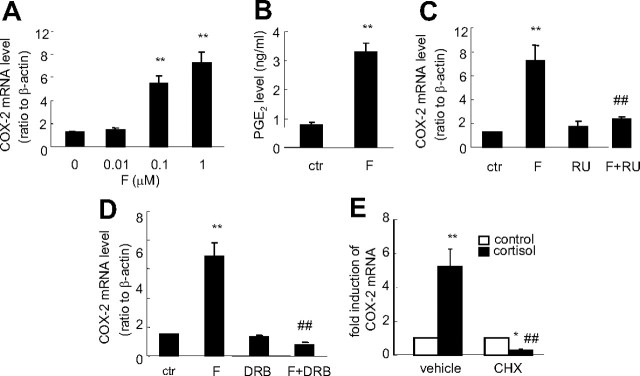
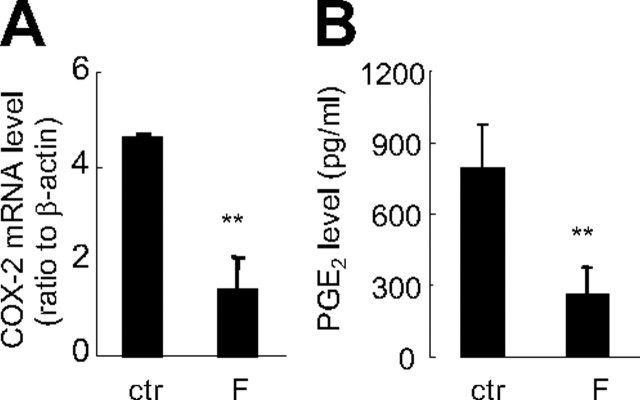
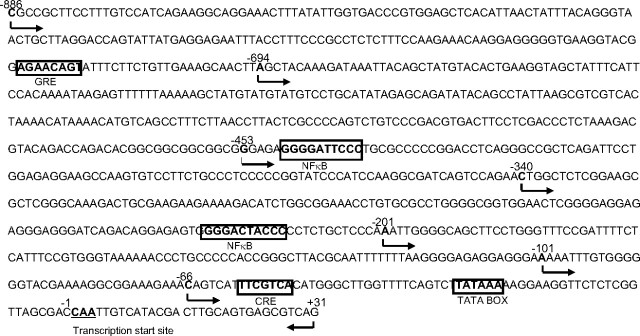
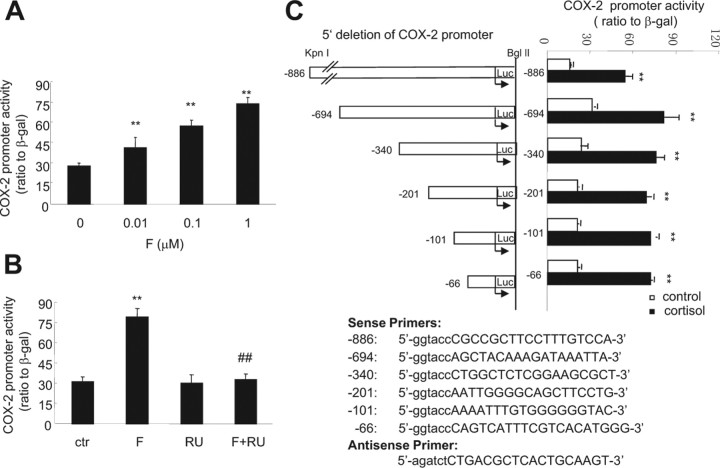
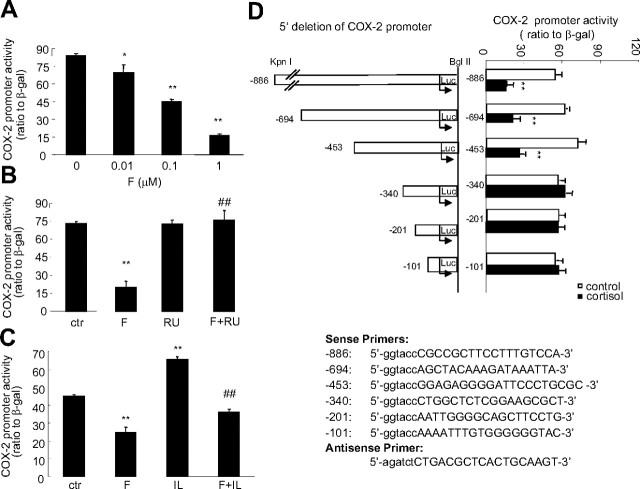
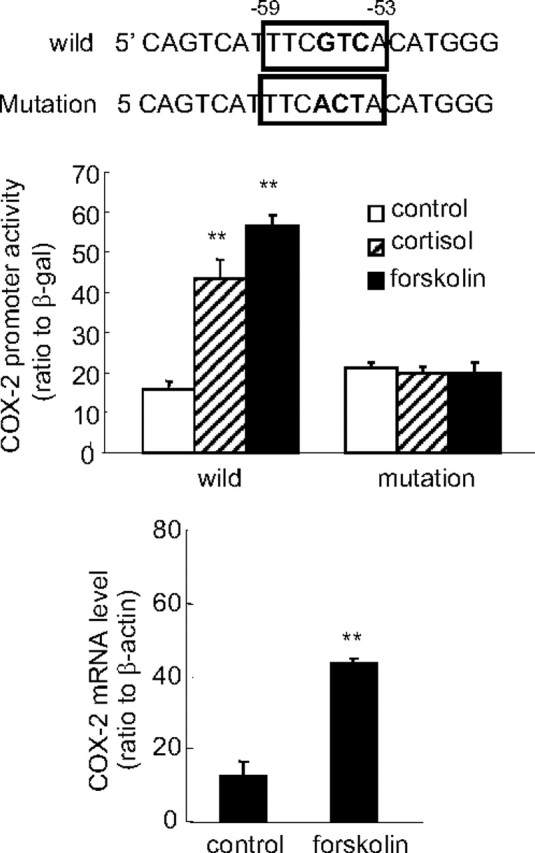
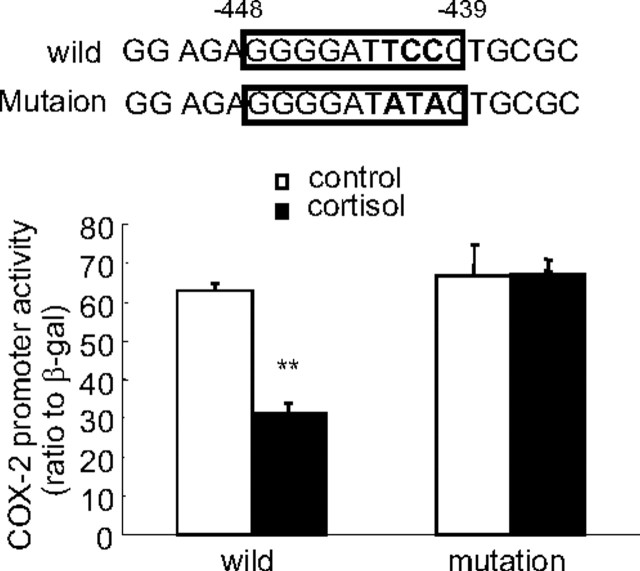
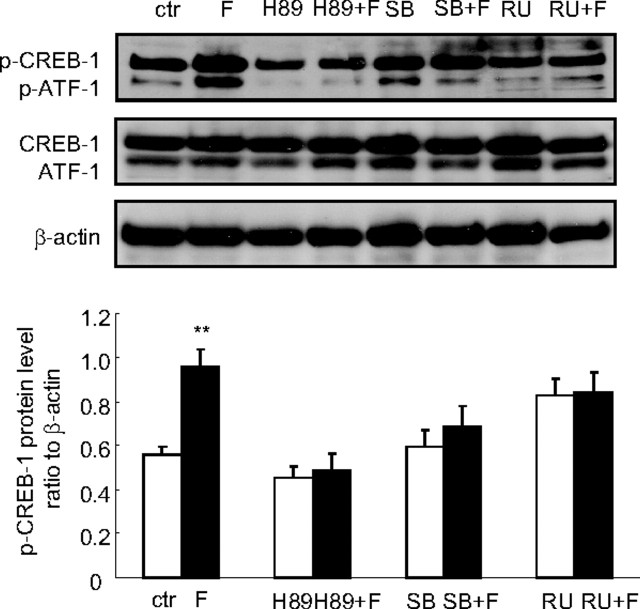
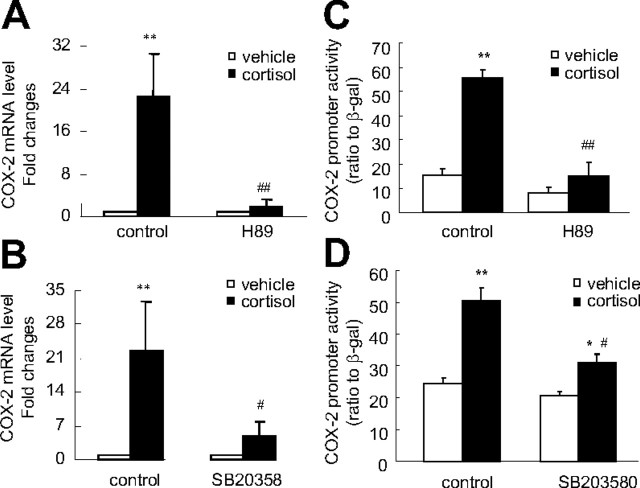
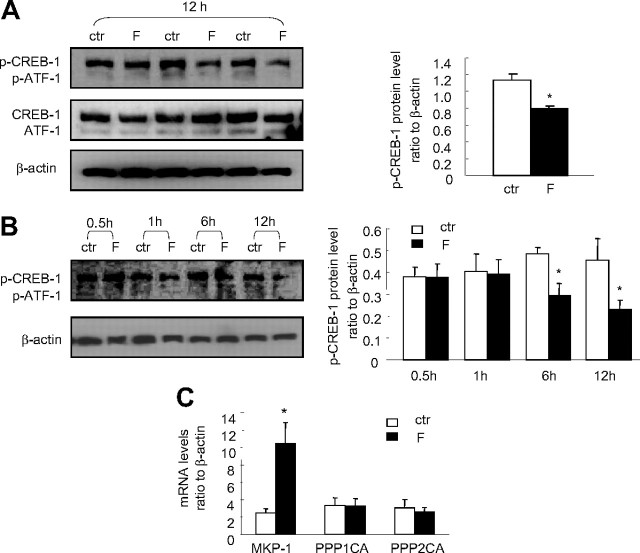
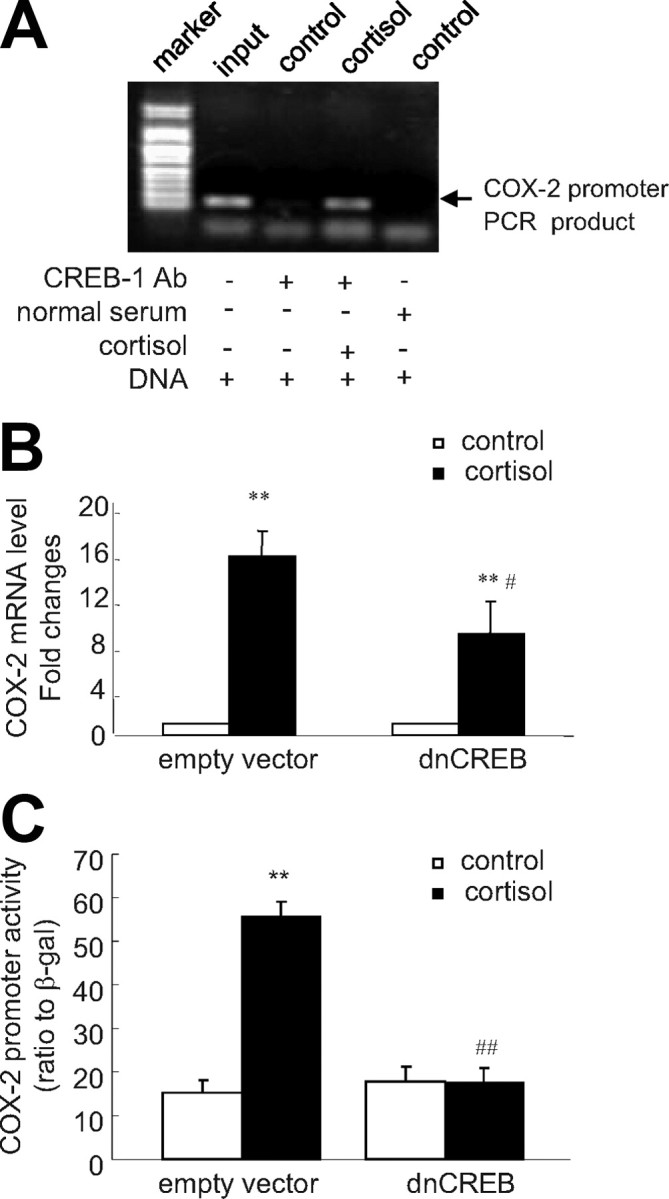
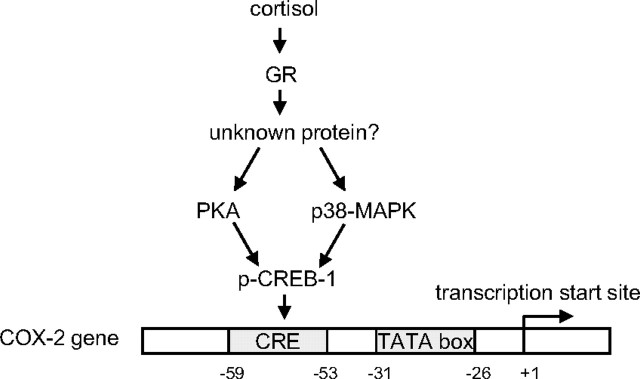
Human amnion fibroblasts produce abundant prostaglandins toward the end of gestation, which is one of the major events leading to parturition. In marked contrast to its well-described antiinflammatory effect, glucocorticoids have been shown to up-regulate cyclooxygenase-2 (COX-2) expression in human amnion fibroblasts. The mechanisms underlying this paradoxical induction of COX-2 by glucocorticoids have not been resolved. Using cultured human amnion fibroblasts, we found that the induction of COX-2 mRNA expression by cortisol was a glucocorticoid receptor (GR)-dependent process requiring ongoing transcription. Upon transfection of a COX-2 promoter-driven reporter gene into the amnion fibroblasts, cortisol stimulated the COX-2 promoter activity. This was abolished by mutagenesis of a cAMP response element (CRE) at -53 to approximately -59bp as well as by cotransfection of a plasmid expressing dominant-negative CRE-binding protein (CREB). The phosphorylation level of CREB-1 was significantly increased by cortisol treatment of the amnion fibroblasts, whereas the effect was attenuated either by the protein kinase A inhibitor H89 or the p38 -MAPK inhibitor SB203580. The induction of the COX-2 promoter activity and the phosphorylation of CREB-1 were also blocked by the GR antagonist RU486. Chromatin immunoprecipitation (ChIP) assay revealed that the binding of CREB-1 to the CRE of the COX-2 promoter was increased by cortisol treatment of the amnion fibroblasts. In conclusion, cortisol, via binding to GR, stimulated COX-2 expression by increasing phosphorylated CREB-1 binding to the CRE of the COX-2 gene. Cortisol may phosphorylate CREB-1 by activating either protein kinase A or p38-MAPK in the amnion fibroblasts.
Figures
Similar articles
-
Cross talk between PKC and CREB in the induction of COX-2 by PGF2α in human amnion fibroblasts.Endocrinology. 2012 Oct;153(10):4938-45. doi: 10.1210/en.2012-1441. Epub 2012 Aug 23. Endocrinology. 2012. PMID: 22919060 Free PMC article.
-
Induction of Galphas contributes to the paradoxical stimulation of cytosolic phospholipase A2alpha expression by cortisol in human amnion fibroblasts.Mol Endocrinol. 2010 May;24(5):1052-61. doi: 10.1210/me.2009-0488. Epub 2010 Mar 4. Mol Endocrinol. 2010. PMID: 20203101 Free PMC article.
-
Role of glucocorticoid receptor and CCAAT/enhancer-binding protein alpha in the feed-forward induction of 11beta-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts.J Endocrinol. 2007 Nov;195(2):241-53. doi: 10.1677/JOE-07-0303. J Endocrinol. 2007. PMID: 17951535
-
Activation of prostaglandin EP4 receptor attenuates the induction of cyclooxygenase-2 expression by EP2 receptor activation in human amnion fibroblasts: implications for parturition.FASEB J. 2019 Jul;33(7):8148-8160. doi: 10.1096/fj.201802642R. Epub 2019 Mar 27. FASEB J. 2019. PMID: 30917001
-
AKAP95-mediated nuclear anchoring of PKA mediates cortisol-induced PTGS2 expression in human amnion fibroblasts.Sci Signal. 2017 Nov 21;10(506):eaac6160. doi: 10.1126/scisignal.aac6160. Sci Signal. 2017. PMID: 29162743
Cited by
-
Preterm Birth: A Narrative Review of the Current Evidence on Nutritional and Bioactive Solutions for Risk Reduction.Nutrients. 2019 Aug 6;11(8):1811. doi: 10.3390/nu11081811. Nutrients. 2019. PMID: 31390765 Free PMC article. Review.
-
Cross talk between PKC and CREB in the induction of COX-2 by PGF2α in human amnion fibroblasts.Endocrinology. 2012 Oct;153(10):4938-45. doi: 10.1210/en.2012-1441. Epub 2012 Aug 23. Endocrinology. 2012. PMID: 22919060 Free PMC article.
-
The dual role of glucocorticoid regeneration in inflammation at parturition.Front Immunol. 2024 Sep 3;15:1459489. doi: 10.3389/fimmu.2024.1459489. eCollection 2024. Front Immunol. 2024. PMID: 39290694 Free PMC article.
-
Paradoxical Induction of ALOX15/15B by Cortisol in Human Amnion Fibroblasts: Implications for Inflammatory Responses of the Fetal Membranes at Parturition.Int J Mol Sci. 2023 Jun 29;24(13):10881. doi: 10.3390/ijms241310881. Int J Mol Sci. 2023. PMID: 37446059 Free PMC article.
-
Involvement of GR and p300 in the induction of H6PD by cortisol in human amnion fibroblasts.Endocrinology. 2012 Dec;153(12):5993-6002. doi: 10.1210/en.2012-1531. Epub 2012 Nov 2. Endocrinology. 2012. PMID: 23125313 Free PMC article.
References
-
- Creasy RK1991. Preventing preterm birth. N Engl J Med 325:727–729 - PubMed
-
- Saigal S, Doyle LW2008. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet 371:261–269 - PubMed
-
- Goldkrand JW, Schulte RL, Messer RH1976. Maternal and fetal plasma cortisol levels at parturition. Obstet Gynecol 47:41–45 - PubMed
-
- Sippell WG, Müller-Holve W, Dörr HG, Bidlingmaier F, Knorr D1981. Concentrations of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, 11-deoxycortisol, cortisol, and cortisone determined simultaneously in human amniotic fluid throughout gestation. J Clin Endocrinol Metab 52:385–392 - PubMed
-
- Whittle WL, Patel FA, Alfaidy N, Holloway AC, Fraser M, Gyomorey S, Lye SJ, Gibb W, Challis JR2001. Glucocorticoid regulation of human and ovine parturition: the relationship between fetal hypothalamic-pituitary-adrenal axis activation and intrauterine prostaglandin production. Biol Reprod 64:1019–1032 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
