

Unsupervised statistical clustering of environmental shotgun sequences
- PMID: 19799776
- PMCID: PMC2765972
- DOI: 10.1186/1471-2105-10-316
Unsupervised statistical clustering of environmental shotgun sequences
Abstract
Background: The development of effective environmental shotgun sequence binning methods remains an ongoing challenge in algorithmic analysis of metagenomic data. While previous methods have focused primarily on supervised learning involving extrinsic data, a first-principles statistical model combined with a self-training fitting method has not yet been developed.
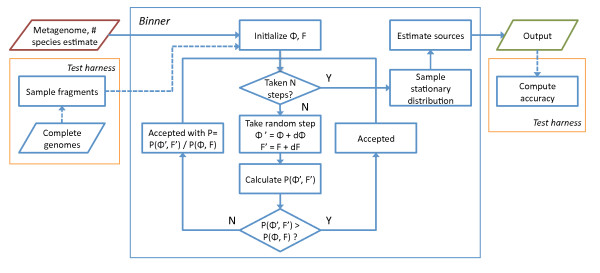
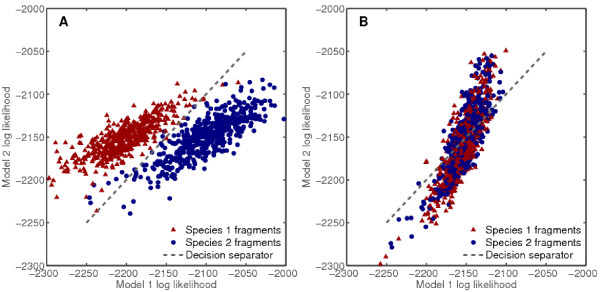
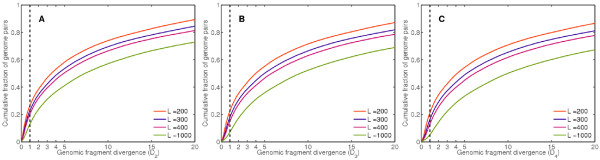
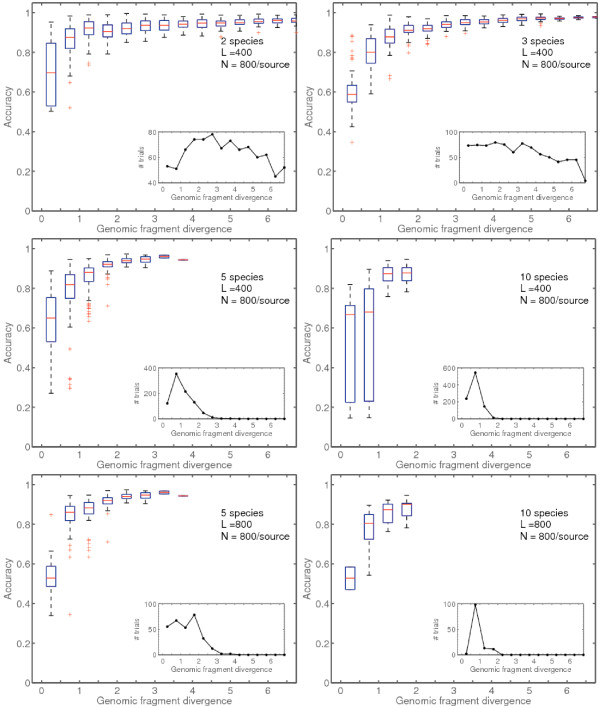
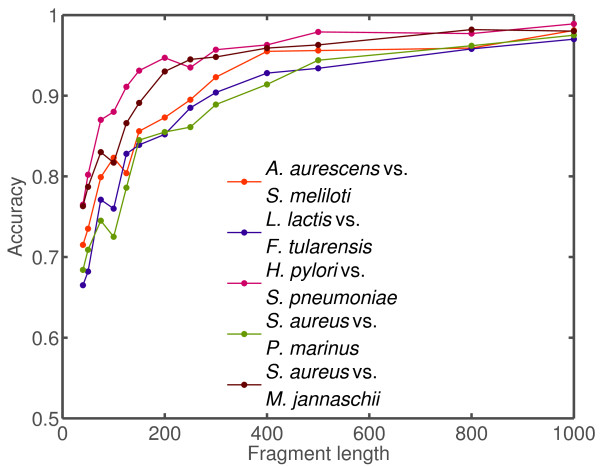
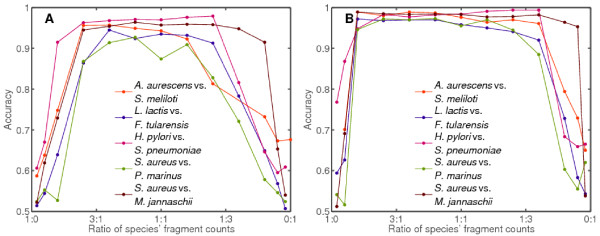
Results: We derive an unsupervised, maximum-likelihood formalism for clustering short sequences by their taxonomic origin on the basis of their k-mer distributions. The formalism is implemented using a Markov Chain Monte Carlo approach in a k-mer feature space. We introduce a space transformation that reduces the dimensionality of the feature space and a genomic fragment divergence measure that strongly correlates with the method's performance. Pairwise analysis of over 1000 completely sequenced genomes reveals that the vast majority of genomes have sufficient genomic fragment divergence to be amenable for binning using the present formalism. Using a high-performance implementation, the binner is able to classify fragments as short as 400 nt with accuracy over 90% in simulations of low-complexity communities of 2 to 10 species, given sufficient genomic fragment divergence. The method is available as an open source package called LikelyBin.
Conclusion: An unsupervised binning method based on statistical signatures of short environmental sequences is a viable stand-alone binning method for low complexity samples. For medium and high complexity samples, we discuss the possibility of combining the current method with other methods as part of an iterative process to enhance the resolving power of sorting reads into taxonomic and/or functional bins.
Figures
References
-
- Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu D, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcón LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter CJ. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology. 2007;5:e77. doi: 10.1371/journal.pbio.0050077. - DOI - PMC - PubMed
-
- Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, Mchardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernández M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
