

On the mechanism underlying (23S)-25-dehydro-1alpha(OH)-vitamin D3-26,23-lactone antagonism of hVDRwt gene activation and its switch to a superagonist
- PMID: 19801650
- PMCID: PMC2794745
- DOI: 10.1074/jbc.M109.042069
On the mechanism underlying (23S)-25-dehydro-1alpha(OH)-vitamin D3-26,23-lactone antagonism of hVDRwt gene activation and its switch to a superagonist
Abstract
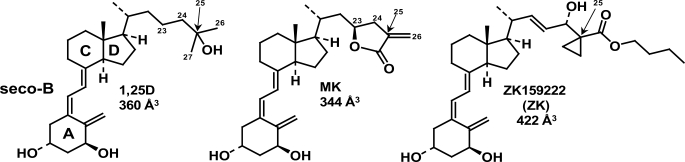
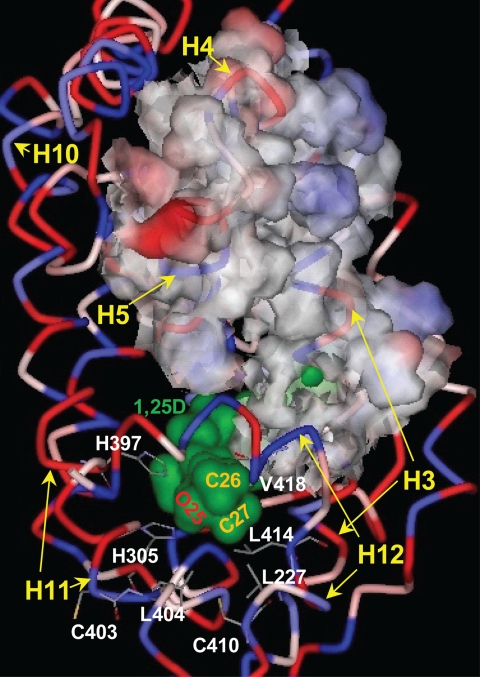
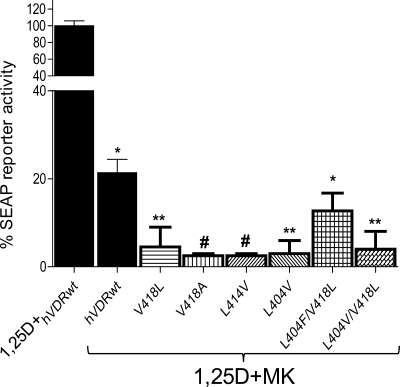
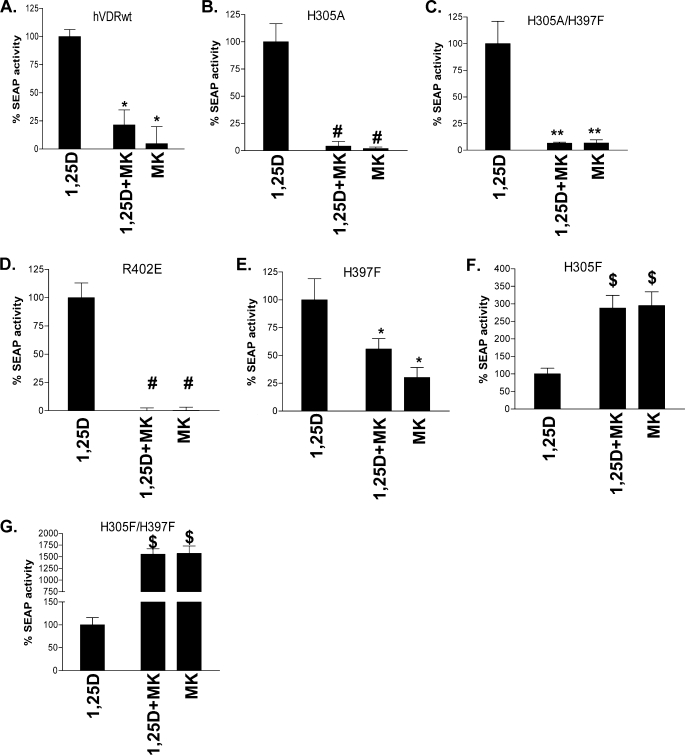
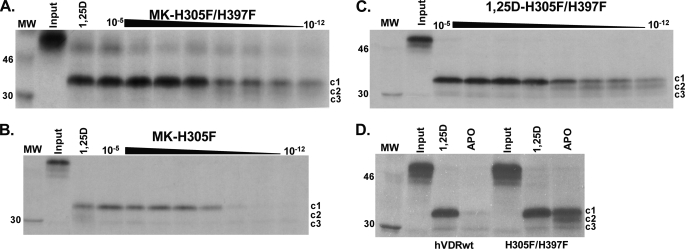
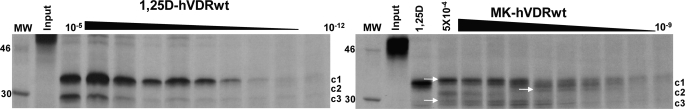
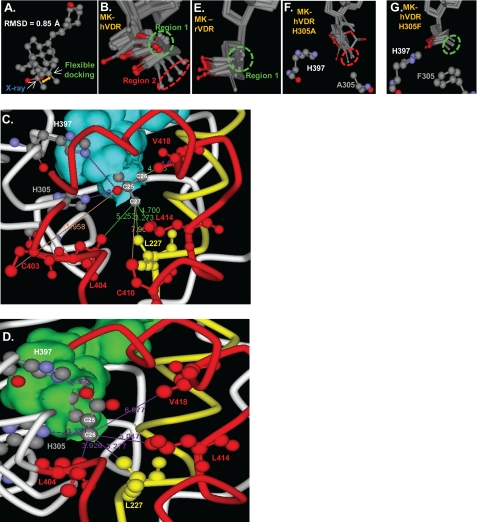
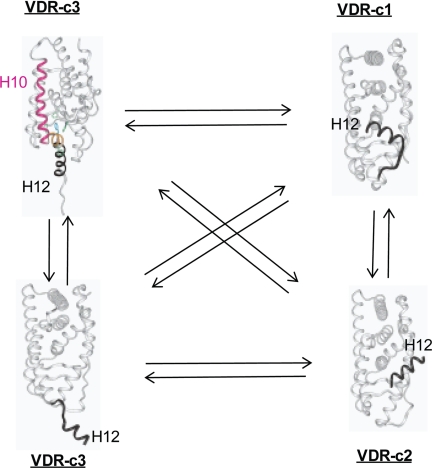
(23S)-25-Dehydro-1alpha(OH)-vitamin D(3)-26,23-lactone (MK) is an antagonist of the 1alpha,25(OH)(2)-vitamin D(3) (1,25D)/human nuclear vitamin D receptor (hVDR) transcription initiation complex, where the activation helix (i.e. helix-12) is closed. To study the mode of antagonism of MK an hVDR mutant library was designed to alter the free molecular volume in the region of the hVDR ligand binding pocket occupied by the ligand side-chain atoms (i.e. proximal to helix-12). The 1,25D-hVDR structure-function studies demonstrate that 1) van der Waals contacts between helix-12 residues Leu-414 and Val-418 and 1,25D enhance the stability of the closed helix-12 conformer and 2) removal of the side-chain H-bonds to His-305(F) and/or His-397(F) have no effect on 1,25D transactivation, even though they reduce the binding affinity of 1,25D. The MK structure-function results demonstrate that the His-305, Leu-404, Leu-414, and Val-418 mutations, which increase the free volume of the hVDR ligand binding pocket, significantly enhance MK antagonist potency. Surprisingly, the H305F and H305F/H397F mutations turn MK into a VDR superagonist (EC(50) approximately 0.05 nm) but do not concomitantly alter MK binding affinity. Molecular modeling studies demonstrate that MK antagonism stems from its side chain energetically preferring a pose in the VDR ligand binding pocket where its terminal C26-methylene atom is far removed from helix-12. MK superagonism results from an energetically favored increase in interaction between Leu-404/Val-418 and C26, resulting in an increase in the stability and population of the closed, helix-12 conformer. Finally, the results/model generated, coupled with application of a VDR ensemble allosterics model, provide an understanding for the species specificity of MK.
Figures
Similar articles
-
Agonist and antagonist binding to the nuclear vitamin D receptor: dynamics, mutation effects and functional implications.In Silico Pharmacol. 2013 Feb 12;1:2. doi: 10.1186/2193-9616-1-2. eCollection 2013. In Silico Pharmacol. 2013. PMID: 25505647 Free PMC article.
-
Structural basis of the histidine-mediated vitamin D receptor agonistic and antagonistic mechanisms of (23S)-25-dehydro-1alpha-hydroxyvitamin D3-26,23-lactone.Acta Crystallogr D Biol Crystallogr. 2010 Aug;66(Pt 8):918-26. doi: 10.1107/S0907444910020810. Epub 2010 Jul 10. Acta Crystallogr D Biol Crystallogr. 2010. PMID: 20693691
-
25-Dehydro-1alpha-hydroxyvitamin D3-26,23S-lactone antagonizes the nuclear vitamin D receptor by mediating a unique noncovalent conformational change.Mol Endocrinol. 2000 Nov;14(11):1788-96. doi: 10.1210/mend.14.11.0552. Mol Endocrinol. 2000. PMID: 11075812
-
Molecular basis of the selective activity of vitamin D analogues.J Cell Biochem. 2003 Feb 1;88(2):274-81. doi: 10.1002/jcb.10337. J Cell Biochem. 2003. PMID: 12520526 Review.
-
Structural Studies of Vitamin D Nuclear Receptor Ligand-Binding Properties.Vitam Horm. 2016;100:83-116. doi: 10.1016/bs.vh.2015.10.003. Epub 2015 Nov 27. Vitam Horm. 2016. PMID: 26827949 Review.
Cited by
-
Control of T cell activation by vitamin D.Nat Immunol. 2011 Jan;12(1):3; author reply 3-4. doi: 10.1038/ni0111-3a. Nat Immunol. 2011. PMID: 21169995 No abstract available.
-
Agonist and antagonist binding to the nuclear vitamin D receptor: dynamics, mutation effects and functional implications.In Silico Pharmacol. 2013 Feb 12;1:2. doi: 10.1186/2193-9616-1-2. eCollection 2013. In Silico Pharmacol. 2013. PMID: 25505647 Free PMC article.
-
Inhibitors for the Vitamin D Receptor-Coregulator Interaction.Vitam Horm. 2016;100:45-82. doi: 10.1016/bs.vh.2015.10.002. Epub 2015 Nov 30. Vitam Horm. 2016. PMID: 26827948 Free PMC article. Review.
-
Vitamin D receptor (VDR) regulation of voltage-gated chloride channels by ligands preferring a VDR-alternative pocket (VDR-AP).Mol Endocrinol. 2011 Aug;25(8):1289-300. doi: 10.1210/me.2010-0442. Epub 2011 Jun 9. Mol Endocrinol. 2011. PMID: 21659475 Free PMC article.
-
Evidence of Allosteric Coupling between Substrate Binding and Adx Recognition in the Vitamin D Carbon-24 Hydroxylase CYP24A1.Biochemistry. 2020 Apr 21;59(15):1537-1548. doi: 10.1021/acs.biochem.0c00107. Epub 2020 Apr 13. Biochemistry. 2020. PMID: 32259445 Free PMC article.
References
-
- Sutton A. L., MacDonald P. N. (2003) Mol. Endocrinol. 17, 777–791 - PubMed
-
- Barletta F., Freedman L. P., Christakos S. (2002) Mol. Endocrinol. 16, 301–314 - PubMed
-
- Rachez C., Freedman L. P. (2000) Gene 246, 9–21 - PubMed
-
- Tocchini-Valentini G., Rochel N., Wurtz J. M., Moras D. (2004) J. Med. Chem. 47, 1956–1961 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
