

RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1
- PMID: 19801658
- PMCID: PMC2787369
- DOI: 10.1074/jbc.M109.040774
RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1
Abstract
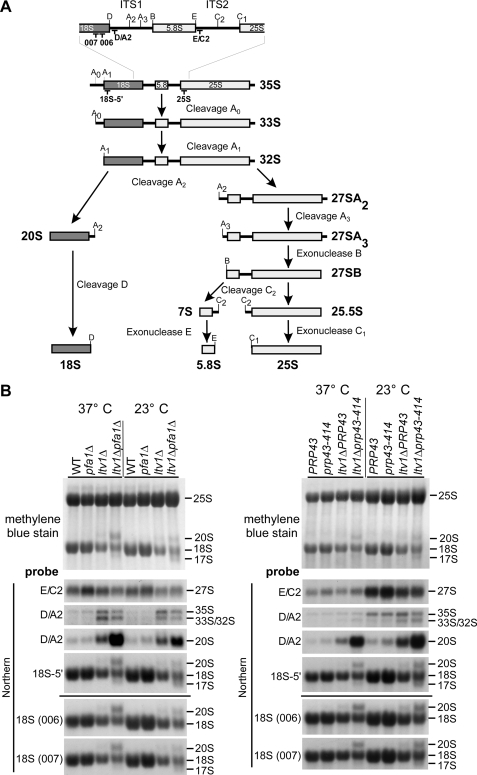
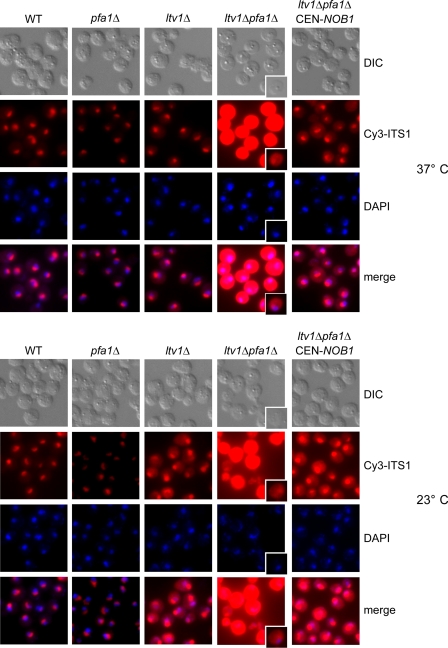
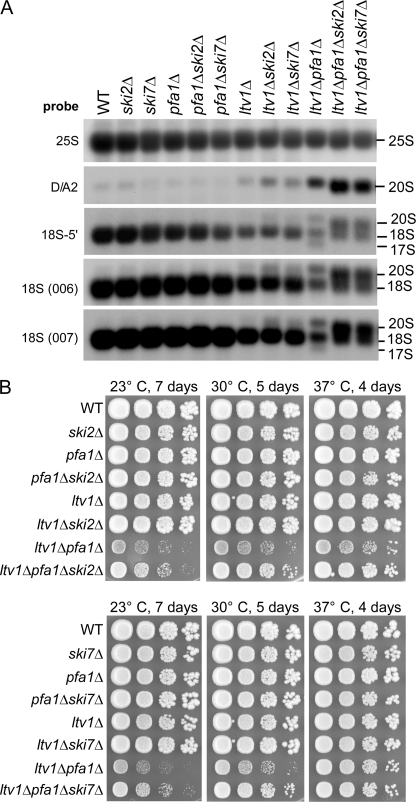
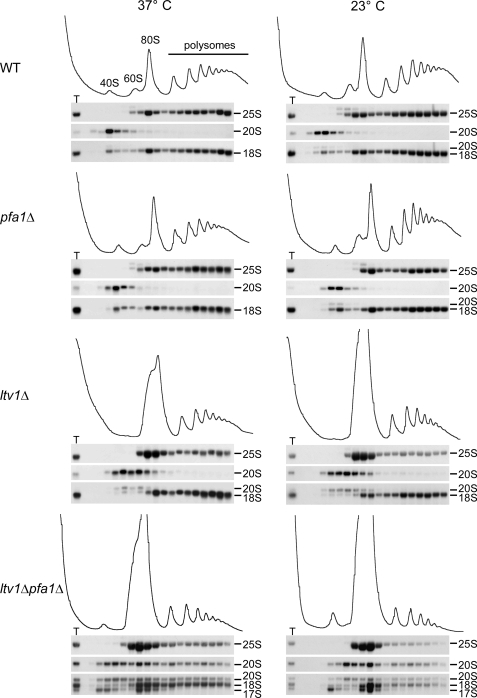
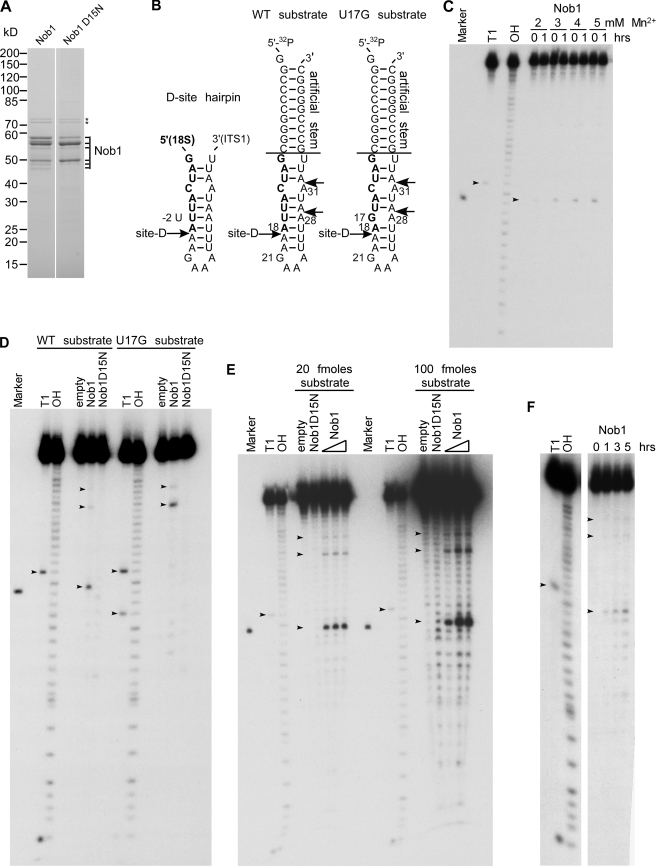
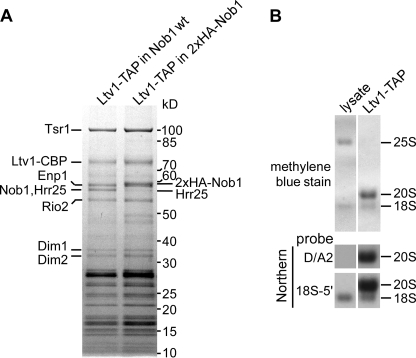
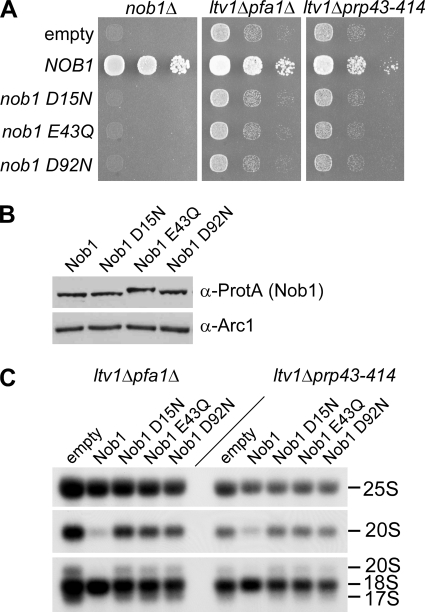
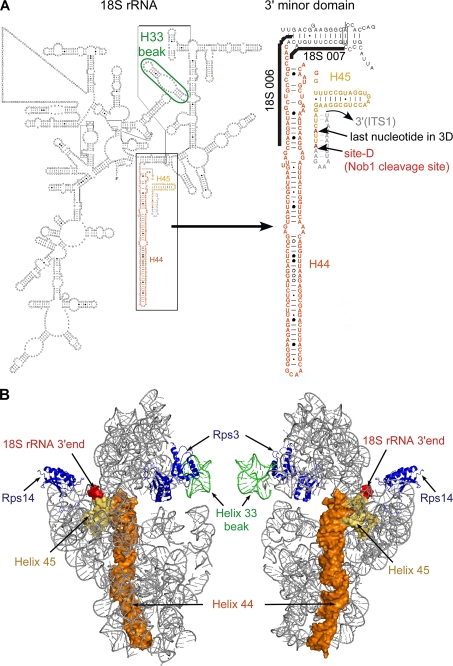
Many RNA nucleases and helicases participate in ribosome biogenesis, but how they cooperate with each other is largely unknown. Here we report that in vivo cleavage of the yeast pre-rRNA at site D, the 3'-end of the 18 S rRNA, requires functional interactions between PIN (PilT N terminus) domain protein Nob1 and the DEAH box RNA helicase Prp43. Nob1 showed specific cleavage on a D-site substrate analogue in vitro, which was abolished by mutations in the Nob1 PIN domain or the RNA substrate. Genetic analyses linked Nob1 to the late pre-40 S-associated factor Ltv1, the RNA helicase Prp43, and its cofactor Pfa1. In strains lacking Ltv1, mutation of Prp43 or Pfa1 led to a striking accumulation of 20 S pre-rRNA in the cytoplasm due to inhibition of site D cleavage. This phenotype was suppressed by increased dosage of wild-type Nob1 but not by Nob1 variants mutated in the catalytic site. In ltv1/pfa1 mutants the 20 S pre-rRNA was susceptible to 3' to 5' degradation by the cytoplasmic exosome. This degraded into the 3' region of the 18 S rRNA, strongly indicating that the preribosomes are structurally defective.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
