

Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11
- PMID: 19804848
- PMCID: PMC2756548
- DOI: 10.1016/j.ajhg.2009.09.005
Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11
Abstract
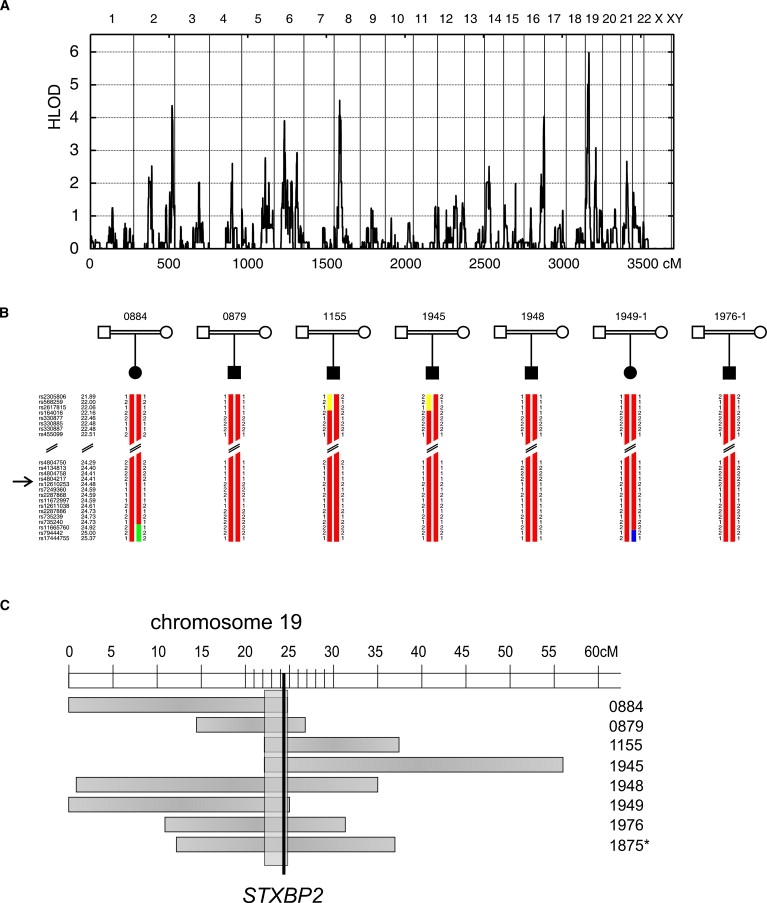
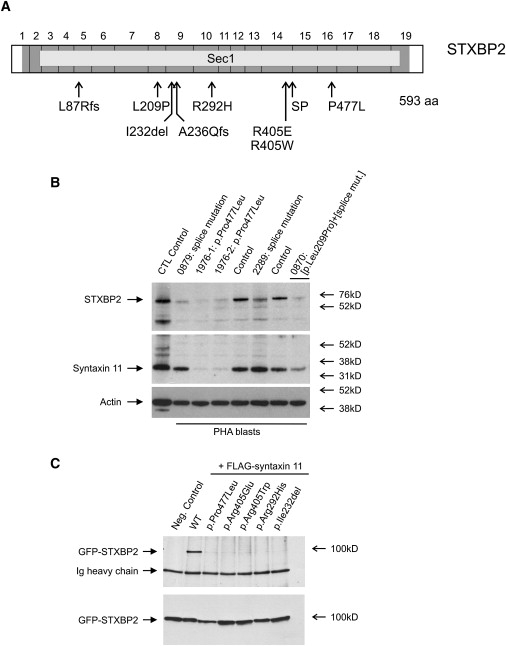
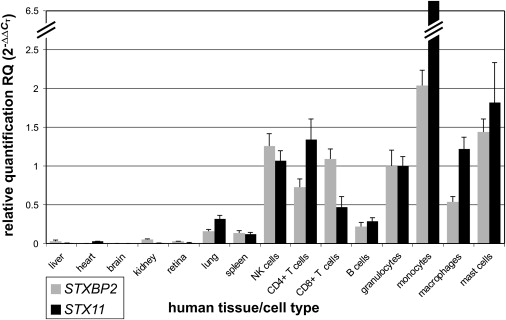
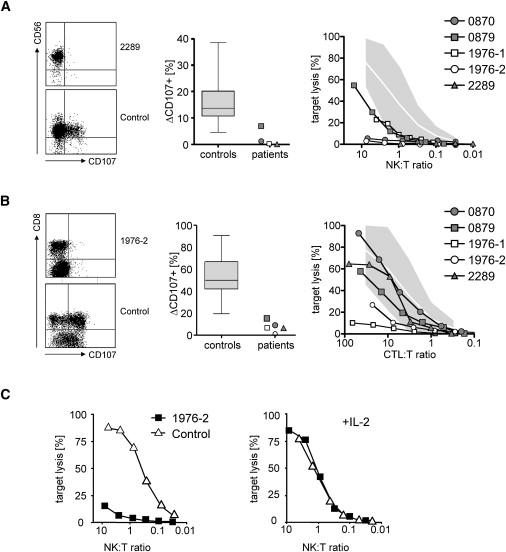
Rapid intracellular transport and secretion of cytotoxic granules through the immunological synapse requires a balanced interaction of several proteins. Disturbance of this highly regulated process underlies familial hemophagocytic lymphohistiocytosis (FHL), a genetically heterogeneous autosomal-recessive disorder characterized by a severe hyperinflammatory phenotype. Here, we have assigned FHL-5 to a 1 Mb region on chromosome 19p by using high-resolution SNP genotyping in eight unrelated FHL patients from consanguineous families. Subsequently, we found nine different mutations, either truncating or missense, in STXBP2 in twelve patients from Turkey, Saudi Arabia, and Central Europe. STXBP2 encodes syntaxin binding protein 2 (Munc18-2), involved in the regulation of vesicle transport to the plasma membrane. We have identified syntaxin 11, a SNARE protein mutated in FHL-4, as an interaction partner of STXBP2. This interaction is eliminated by the missense mutations found in our FHL-5 patients, which leads to a decreased stability of both proteins, as shown in patient lymphocytes. Activity of natural killer and cytotoxic T cells was markedly reduced or absent, as determined by CD107 degranulation. Our findings thus identify a key role for STXBP2 in lytic granule exocytosis.
Figures
References
-
- Henter J.I., Samuelsson-Horne A., Arico M., Egeler R.M., Elinder G., Filipovich A.H., Gadner H., Imashuku S., Komp D., Ladisch S. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100:2367–2373. - PubMed
-
- Stephan J.L., Donadieu J., Ledeist F., Blanche S., Griscelli C., Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82:2319–2323. - PubMed
-
- Arico M., Janka G., Fischer A., Henter J.I., Blanche S., Elinder G., Martinetti M., Rusca M.P. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10:197–203. - PubMed
-
- Janka G.E. Familial hemophagocytic lymphohistiocytosis. Eur. J. Pediatr. 1983;140:221–230. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
