

Insulin resistance is a cellular antioxidant defense mechanism
- PMID: 19805130
- PMCID: PMC2764908
- DOI: 10.1073/pnas.0902380106
Insulin resistance is a cellular antioxidant defense mechanism
Abstract
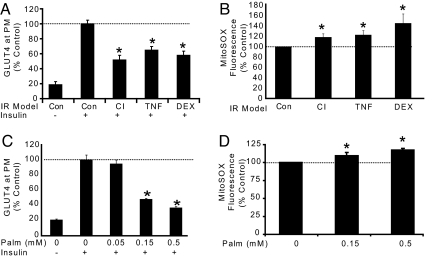
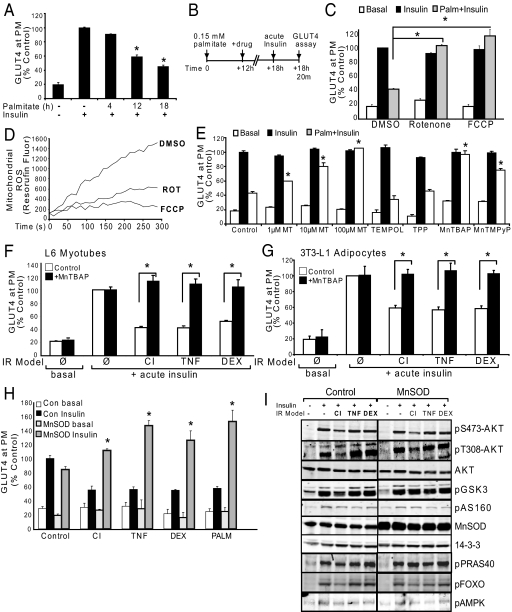
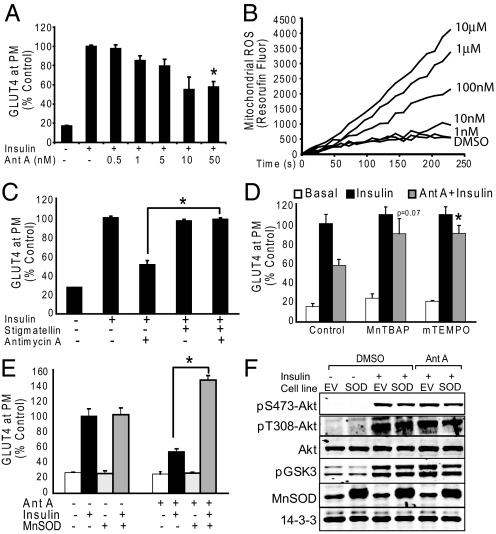
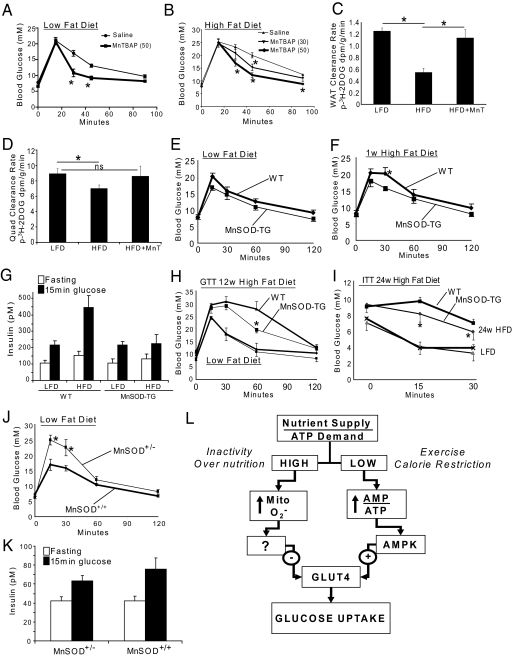
We know a great deal about the cellular response to starvation via AMPK, but less is known about the reaction to nutrient excess. Insulin resistance may be an appropriate response to nutrient excess, but the cellular sensors that link these parameters remain poorly defined. In the present study we provide evidence that mitochondrial superoxide production is a common feature of many different models of insulin resistance in adipocytes, myotubes, and mice. In particular, insulin resistance was rapidly reversible upon exposure to agents that act as mitochondrial uncouplers, ETC inhibitors, or mitochondrial superoxide dismutase (MnSOD) mimetics. Similar effects were observed with overexpression of mitochondrial MnSOD. Furthermore, acute induction of mitochondrial superoxide production using the complex III antagonist antimycin A caused rapid attenuation of insulin action independently of changes in the canonical PI3K/Akt pathway. These results were validated in vivo in that MnSOD transgenic mice were partially protected against HFD induced insulin resistance and MnSOD+/- mice were glucose intolerant on a standard chow diet. These data place mitochondrial superoxide at the nexus between intracellular metabolism and the control of insulin action potentially defining this as a metabolic sensor of energy excess.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Holland WL, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. - PubMed
-
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: Insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
