

Enzyme millisecond conformational dynamics do not catalyze the chemical step
- PMID: 19805169
- PMCID: PMC2762662
- DOI: 10.1073/pnas.0909150106
Enzyme millisecond conformational dynamics do not catalyze the chemical step
Abstract
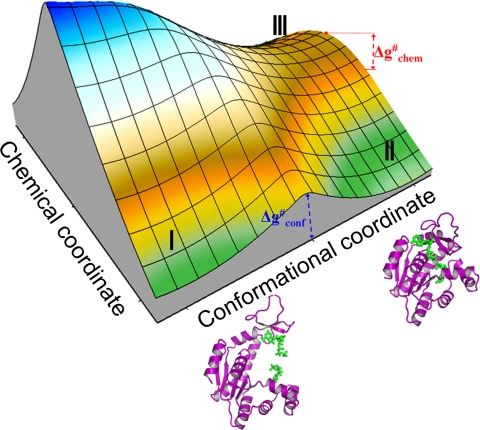
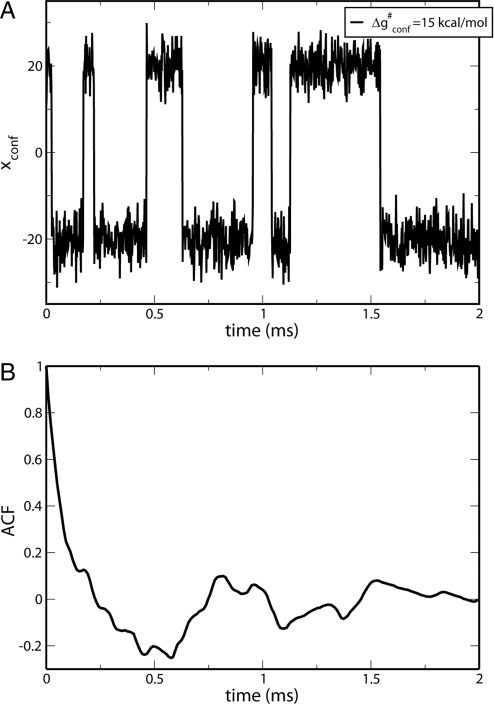
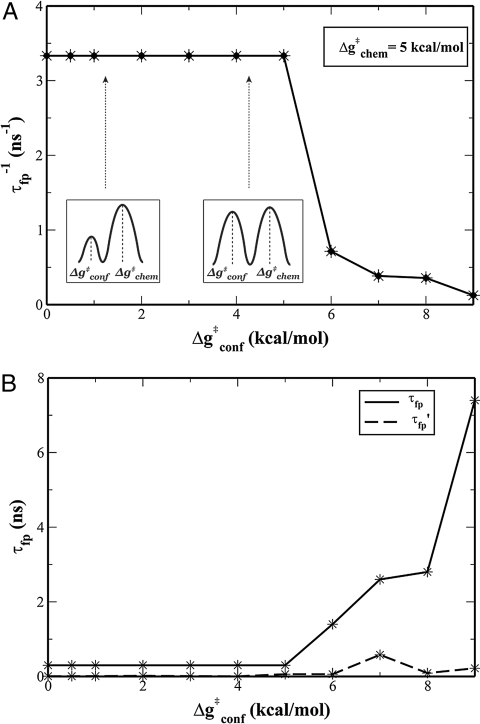
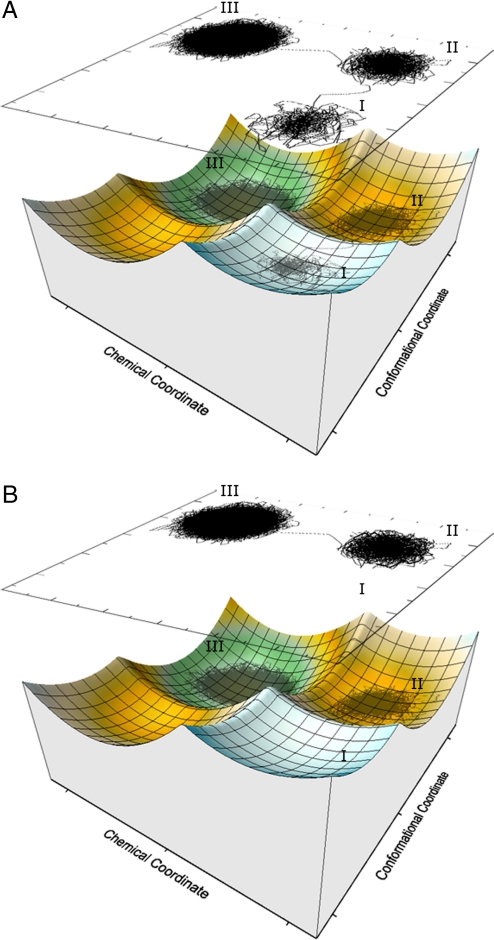
The idea that enzymes catalyze reactions by dynamical coupling between the conformational motions and the chemical coordinates has recently attracted major experimental and theoretical interest. However, experimental studies have not directly established that the conformational motions transfer energy to the chemical coordinate, and simulating enzyme catalysis on the relevant timescales has been impractical. Here, we introduce a renormalization approach that transforms the energetics and dynamics of the enzyme to an equivalent low-dimensional system, and allows us to simulate the dynamical coupling on a ms timescale. The simulations establish, by means of several independent approaches, that the conformational dynamics is not remembered during the chemical step and does not contribute significantly to catalysis. Nevertheless, the precise nature of this coupling is a question of great importance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
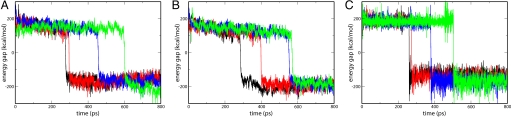
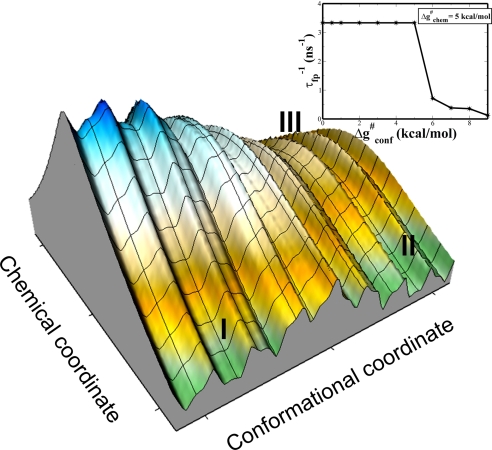
Comment in
-
Role of conformation transitions in adenylate kinase.Proc Natl Acad Sci U S A. 2010 Apr 27;107(17):E71; author reply E72. doi: 10.1073/pnas.1002180107. Proc Natl Acad Sci U S A. 2010. PMID: 20424124 Free PMC article. No abstract available.
References
-
- Warshel A, et al. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106:3210–3235. - PubMed
-
- Hansson T, Nordlund P, Åqvist J. Energetics of nucleophile activation in a protein tyrosine phosphatase. J Mol Biol. 1997;265:118–127. - PubMed
-
- Akke M. Out of hot water. Nat Struct Mol Biol. 2004;11:912–913. - PubMed
-
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. - PubMed
-
- Eisenmesser EZ, et al. Intrinsic dynamcs of an enzyme underlies catalysis. Nature. 2005;438:117–121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
