

Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant
- PMID: 19805308
- PMCID: PMC2752539
- DOI: 10.1073/pnas.0906784106
Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant
Abstract
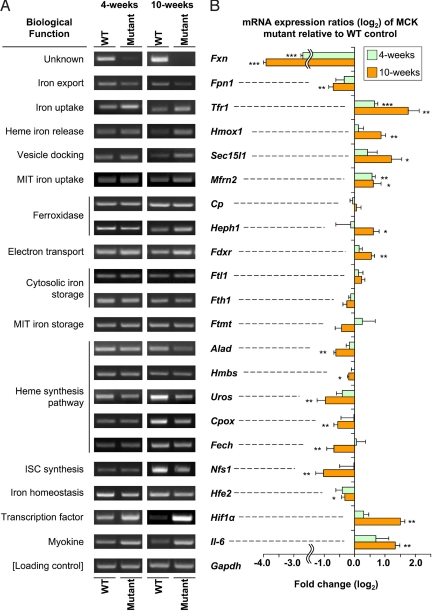
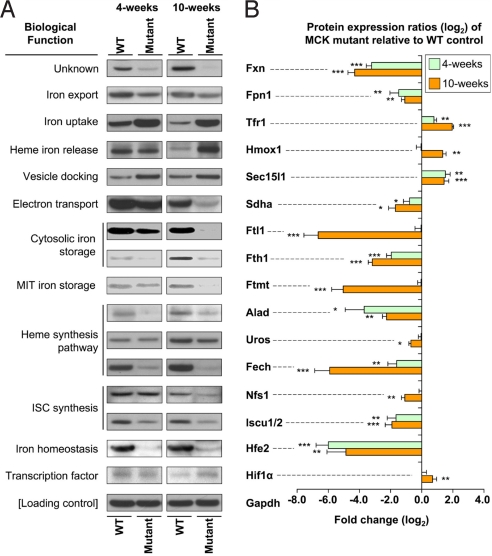
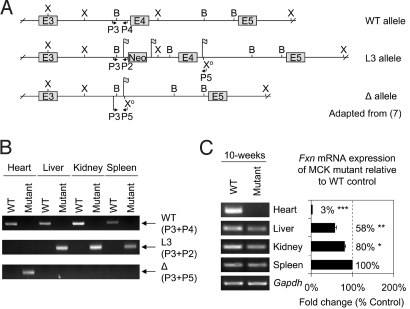
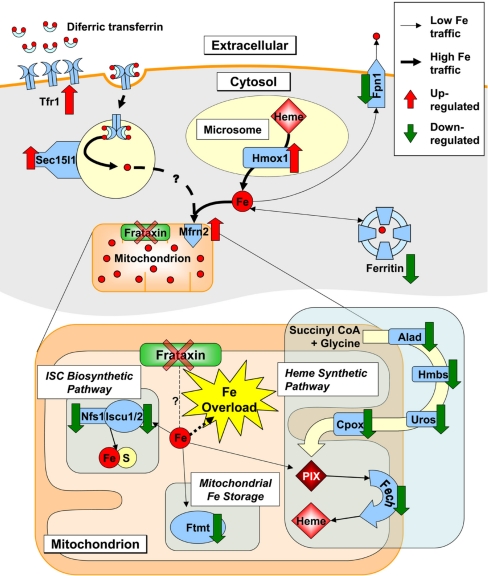
We used the muscle creatine kinase (MCK) conditional frataxin knockout mouse to elucidate how frataxin deficiency alters iron metabolism. This is of significance because frataxin deficiency leads to Friedreich's ataxia, a disease marked by neurologic and cardiologic degeneration. Using cardiac tissues, we demonstrate that frataxin deficiency leads to down-regulation of key molecules involved in 3 mitochondrial utilization pathways: iron-sulfur cluster (ISC) synthesis (iron-sulfur cluster scaffold protein1/2 and the cysteine desulferase Nfs1), mitochondrial iron storage (mitochondrial ferritin), and heme synthesis (5-aminolevulinate dehydratase, coproporphyrinogen oxidase, hydroxymethylbilane synthase, uroporphyrinogen III synthase, and ferrochelatase). This marked decrease in mitochondrial iron utilization and resultant reduced release of heme and ISC from the mitochondrion could contribute to the excessive mitochondrial iron observed. This effect is compounded by increased iron availability for mitochondrial uptake through (i) transferrin receptor1 up-regulation, increasing iron uptake from transferrin; (ii) decreased ferroportin1 expression, limiting iron export; (iii) increased expression of the heme catabolism enzyme heme oxygenase1 and down-regulation of ferritin-H and -L, both likely leading to increased "free iron" for mitochondrial uptake; and (iv) increased expression of the mammalian exocyst protein Sec15l1 and the mitochondrial iron importer mitoferrin-2 (Mfrn2), which facilitate cellular iron uptake and mitochondrial iron influx, respectively. Our results enable the construction of a model explaining the cytosolic iron deficiency and mitochondrial iron loading in the absence of frataxin, which is important for understanding the pathogenesis of Friedreich's ataxia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells.Hum Mol Genet. 2005 Dec 15;14(24):3787-99. doi: 10.1093/hmg/ddi393. Epub 2005 Oct 20. Hum Mol Genet. 2005. PMID: 16239244
-
Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia.Proc Natl Acad Sci U S A. 2012 Dec 11;109(50):20590-5. doi: 10.1073/pnas.1215349109. Epub 2012 Nov 20. Proc Natl Acad Sci U S A. 2012. PMID: 23169664 Free PMC article.
-
The ins and outs of mitochondrial iron-loading: the metabolic defect in Friedreich's ataxia.J Mol Med (Berl). 2010 Apr;88(4):323-9. doi: 10.1007/s00109-009-0565-x. Epub 2009 Dec 9. J Mol Med (Berl). 2010. PMID: 19997898 Review.
-
The MCK mouse heart model of Friedreich's ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation.Proc Natl Acad Sci U S A. 2008 Jul 15;105(28):9757-62. doi: 10.1073/pnas.0804261105. Epub 2008 Jul 9. Proc Natl Acad Sci U S A. 2008. PMID: 18621680 Free PMC article.
-
Frataxin and mitochondrial FeS cluster biogenesis.J Biol Chem. 2010 Aug 27;285(35):26737-26743. doi: 10.1074/jbc.R110.118679. Epub 2010 Jun 3. J Biol Chem. 2010. PMID: 20522547 Free PMC article. Review.
Cited by
-
Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease.Biochemistry. 2010 Jun 22;49(24):4945-56. doi: 10.1021/bi1004798. Biochemistry. 2010. PMID: 20481466 Free PMC article. Review.
-
Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol.Proc Natl Acad Sci U S A. 2010 Jun 15;107(24):10775-82. doi: 10.1073/pnas.0912925107. Epub 2010 May 21. Proc Natl Acad Sci U S A. 2010. PMID: 20495089 Free PMC article. Review.
-
Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease.Front Pharmacol. 2014 Mar 3;5:29. doi: 10.3389/fphar.2014.00029. eCollection 2014. Front Pharmacol. 2014. PMID: 24624085 Free PMC article. Review.
-
The "Iron"-y of Iron Overload and Iron Deficiency in Chronic Obstructive Pulmonary Disease.Am J Respir Crit Care Med. 2017 Nov 1;196(9):1103-1112. doi: 10.1164/rccm.201702-0311PP. Am J Respir Crit Care Med. 2017. PMID: 28410559 Free PMC article. No abstract available.
-
Molecular Mechanisms and Therapeutic Targeting of Ferroptosis in Doxorubicin-Induced Cardiotoxicity.JACC Basic Transl Sci. 2024 Jan 3;9(6):811-826. doi: 10.1016/j.jacbts.2023.10.009. eCollection 2024 Jun. JACC Basic Transl Sci. 2024. PMID: 39070280 Free PMC article. Review.
References
-
- Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood. 2005;105:1867–1874. - PubMed
-
- Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAATTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem. 2001;276:27171–27177. - PubMed
-
- Babcock M, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–1712. - PubMed
-
- Rotig A, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. - PubMed
-
- Zhang Y, et al. Mrs3p, Mrs4p, and frataxin provide iron for Fe-S cluster synthesis in mitochondria. J Biol Chem. 2006;281:22493–22502. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
